spettroscopia ad infrarossi
La spettrometria a infrarossi è estremamente utile per le determinazioni qualitative di composti organici e per dedurre strutture molecolari dai loro gruppi funzionali di composti sia organici che inorganici.
Nell'analisi qualitativa la spettroscopia infrarossa può essere utilizzata per l'identificazione di sostanze pure o per l'assorbimento, la localizzazione e l'identificazione di impurità.
Per individuare un'impurità in una sostanza, viene effettuato un confronto tra lo spettro della sostanza studiata e un campione della sostanza pura. Le impurità fanno apparire nello spettro ulteriori bande di assorbimento.
Nell'IR trovano sempre maggiore impiego anche nell'analisi quantitativa, il principale campo di applicazione di questo tipo di analisi è nella quantificazione degli inquinanti atmosferici che provengono da processi industriali.
Una parte dello spettro elettromagnetico che si estende da 0,8 a 1000μm (che corrisponde al numero d'onda compreso tra 12800 e 10 cm-1), è considerata la regione dell'infrarosso che si divide in tre regioni denominate:
a).- IR Vicino. b).- IR fondamentale o medio c).- IR lontano
Ogni tipo di legame assorbe la radiazione infrarossa a una frequenza diversa, il che consente di determinare quale tipo di gruppi funzionali ha la molecola in esame. Gli spettrofotometri a infrarossi lavorano nel medio infrarosso e scansionano da 4000 c m − 1 a 400 c m − 1
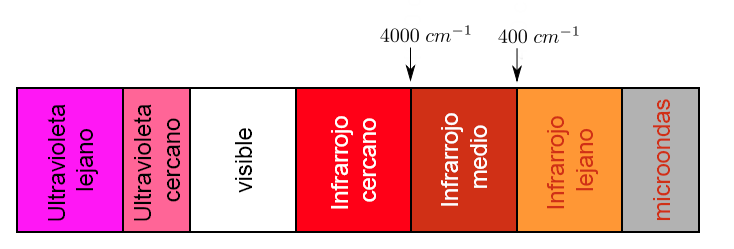
Tuttavia, la regione di importanza analitica è la regione IR fondamentale poiché la maggior parte degli strumenti a infrarossi copre questa regione.
La maggior parte dei materiali organici e inorganici dimostra assorbimento e lo spettro è causato principalmente dallo stiramento e dalla flessione vibrazionale all'interno della molecola. Lo spettro infrarosso è una delle proprietà più caratteristiche di un composto poiché non esistono due spettri identici per due composti diversi, è come un'impronta digitale.
All'interno della regione IR Fondamentale ci sono due regioni, una delle quali è la cosiddetta
i gruppi funzionali da 4000 cm-1 a 1300 cm-1 e la regione delle dita da 1300 cm-1 a 670 cm-1.
Nella regione dei gruppi funzionali, la posizione del picco di assorbimento è più alta o più bassa a seconda solo del gruppo funzionale dove arriva l'assorbimento e non della struttura molecolare completa. La posizione dei picchi nella regione del dito dipende dalla struttura molecolare completa.
Caratteristiche che deve avere una vibrazione per produrre una banda di assorbimento:
La radiazione incidente deve avere una frequenza pari alla frequenza della vibrazione che produrrà.
Che la vibrazione risultante produca un cambiamento nel momento di dipolo, cioè la vibrazione non assorbirà la radiazione infrarossa, se non c'è cambiamento nel momento di dipolo si chiama vibrazione inattiva e saranno attive quando c'è detto cambiamento nel dipolo momento.
Tradizionalmente, l'asse x degli spettri infrarossi utilizza il numero di onde ( ν ¯ , leggi "nu bar"') ed è definito come l'inverso della lunghezza d'onda in cm. ν ¯ = 1 λ . L'asse y rappresenta la percentuale di radiazione trasmessa (trasmittanza) che è rappresentata da % T . Di seguito è mostrata la forma dello spettro infrarosso dell'esano.
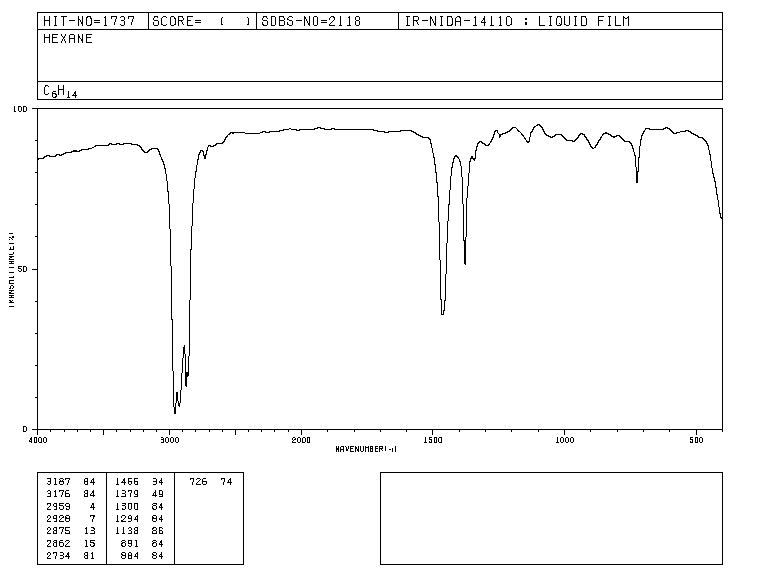
Le bande rappresentano le aree in cui i legami della molecola assorbono la radiazione infrarossa. Nelle bande la trasmittanza è piccola e l'assorbanza è grande.
tipi di vibrazione
Vibrazione di tensione (stretching). Gli atomi tenuti insieme da legami singoli, doppi o tripli si muovono avanti e indietro nella direzione del legame, proprio come oscillano due masse attaccate a una molla.
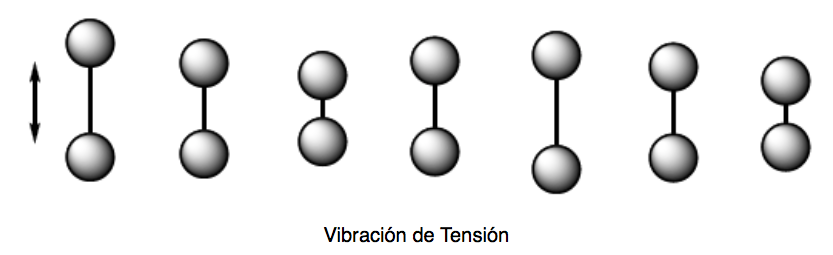
Esistono due modalità di vibrazione della tensione: simmetrica e asimmetrica.
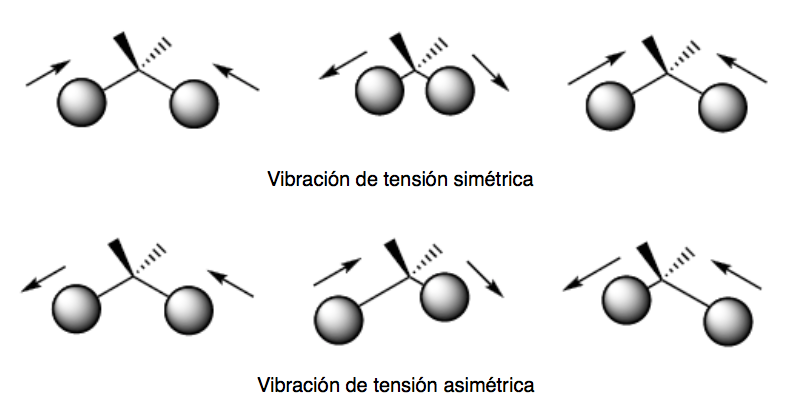
vibrazione di flessione. Gli atomi vibrano in modo che gli angoli varino, ma le lunghezze dei legami no. Esistono quattro modalità di vibrazione della flessione: forbice, oscillazione, scodinzolamento e torsione.
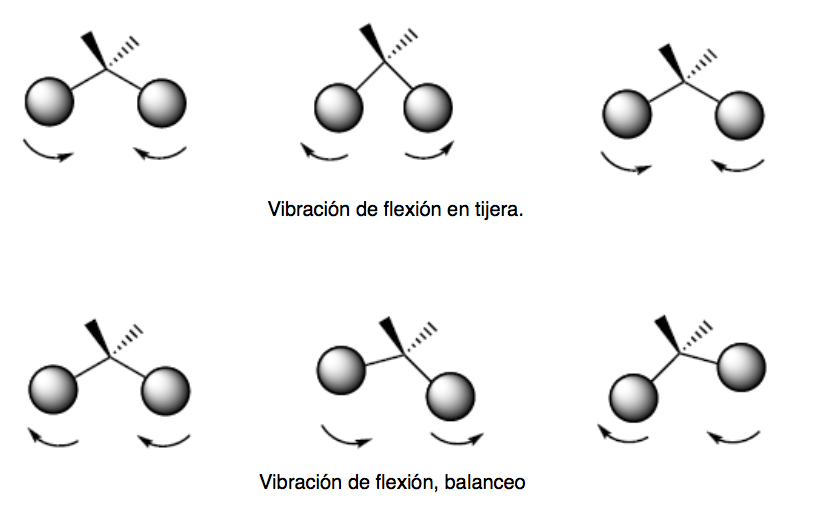
Questi due modi di vibrazione hanno luogo nel piano che contiene i tre atomi che partecipano alla vibrazione.
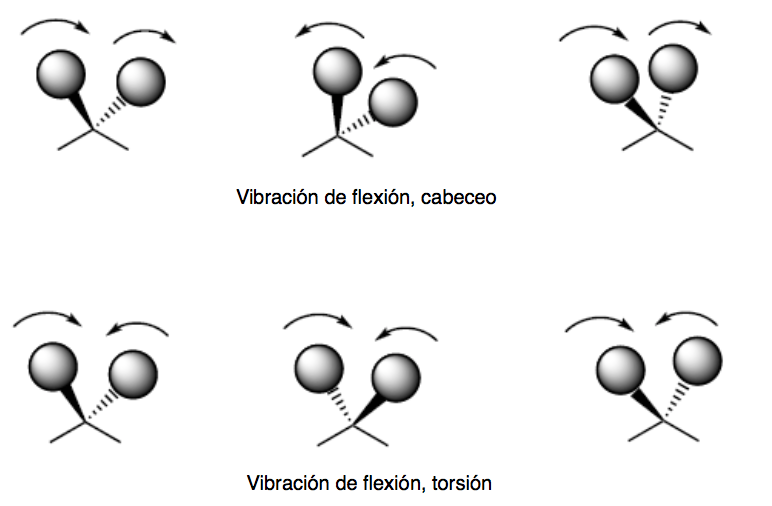
Le modalità di vibrazione del passo e della torsione si verificano fuori dal piano (Fuori dal piano) e sono solitamente rappresentate da Oop.
oscillatore armonico quantistico
Le vibrazioni molecolari possono essere studiate con il modello dell'oscillatore armonico quantistico. L'energia è data da:
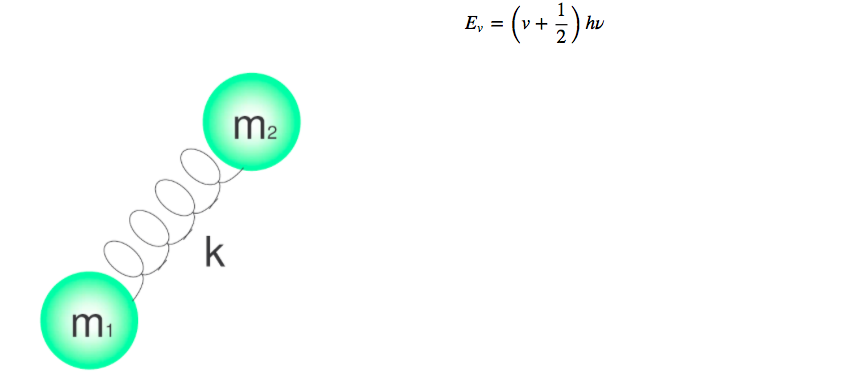
I diversi livelli energetici sono dati dal numero quantico v, che assume valori 0.1.2.3.4.....
h è la costante di Planck e ν è la frequenza dell'oscillatore data dall'espressione:
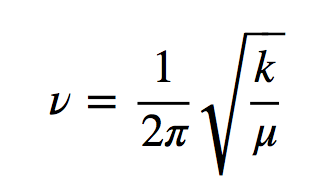
dove k è la forza costante della molla e μ la massa ridotta del sistema
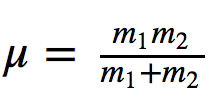
Dividendo la frequenza per la velocità della luce si ottiene il numero di onde ν ¯
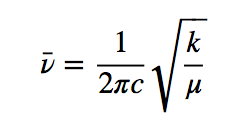
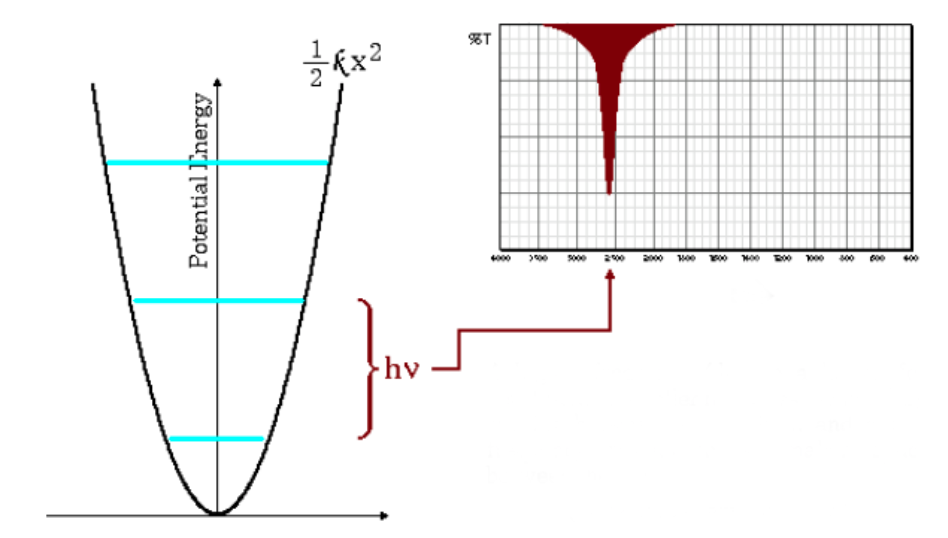
L'equazione di cui sopra indica che piccole masse ridotte (atomi di massa ridotta) e costanti di forza elevate (legami forti) portano ad alte frequenze. In queste condizioni le bande di assorbimento emergono ad alti numeri d'onda.
Come si può vedere nel grafico, le alte frequenze danno luogo a una maggiore spaziatura tra i livelli energetici.
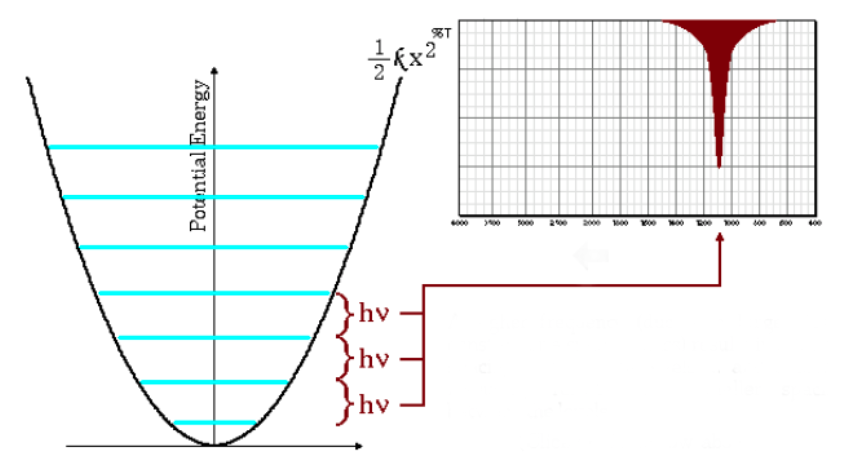
Basse frequenze di assorbimento
Anche l'equazione di cui sopra indica che grandi masse ridotte e piccole costanti di forza (legami deboli) portano a basse frequenze. In queste condizioni le bande di assorbimento escono a basso numero d'onda.
Come si può vedere nel grafico, le basse frequenze danno luogo a una minore spaziatura tra i livelli energetici.
Principali vibrazioni molecolari
Spettro IR
In uno spettro infrarosso, la frequenza (in numero d'onda) viene tracciata rispetto alla percentuale di luce trasmessa (trasmittanza). La percentuale di trasmittanza è definita come il quoziente tra l'intensità della luce trasmessa attraverso il campione, I M , e l'intensità della luce del raggio di riferimento I R moltiplicato per 100.
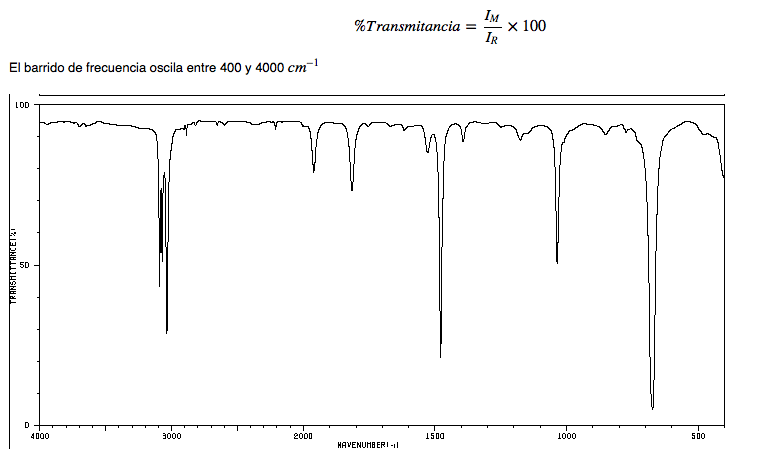
Spettro IR degli alcani
• Deformazione CH: gli alcani mostrano vibrazioni di deformazione CH leggermente inferiori a 3000 c m − 1
• CH bending: i CH 2 della catena mostrano vibrazioni di flessione (a forbice) a 1465 c m − 1 , mentre i metili producono una banda a 1375 c m − 1 dovuta alla vibrazione di flessione simmetrica e un'altra a 1450 c m − 1 dovuta a vibrazione di flessione asimmetrica. Tutte le bande push-up sono di media intensità.
Si noti che la banda di flessione asimmetrica del metile si sovrappone alla banda di flessione a forbice di CH 2 .
Spettro IR dell'esano
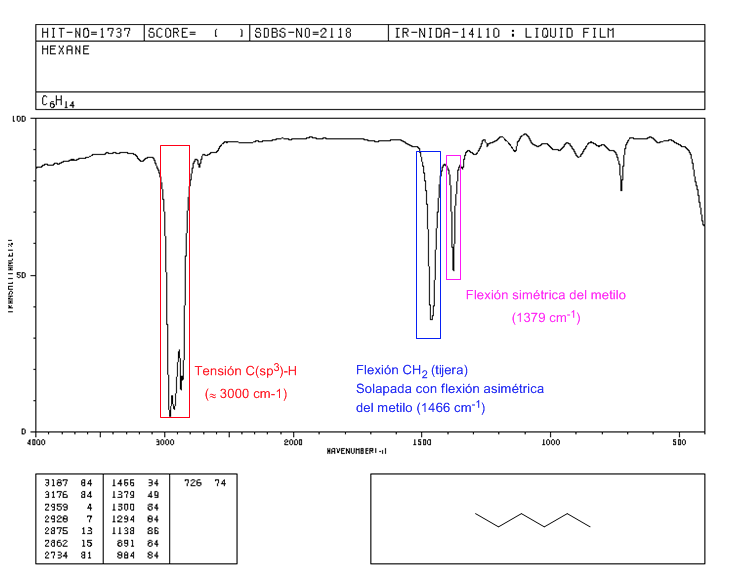
Spettro del 2,2-dimetilbutano
La presenza del gruppo tert-butile produce la scissione della banda di flessione simmetrica in due bande a 1390 e 1370 c m − 1 . La banda al 1390 è forte la metà di quella al 1370.
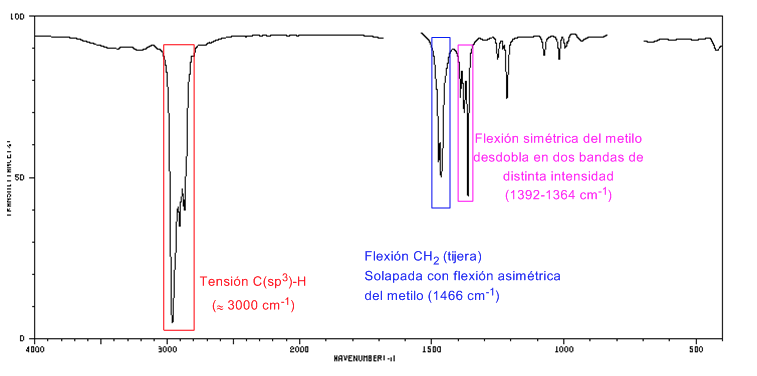
Spettro IR dei cicloalcani
I cicloalcani hanno uno spettro IR molto simile agli alcani con una banda di stiramento CH leggermente inferiore a 3000 c m − 1 e una banda di flessione CH a forbice per CH 2 a 1465 c m − 1 . La principale differenza con gli alcani è l'assenza della banda elastica simmetrica del metile. 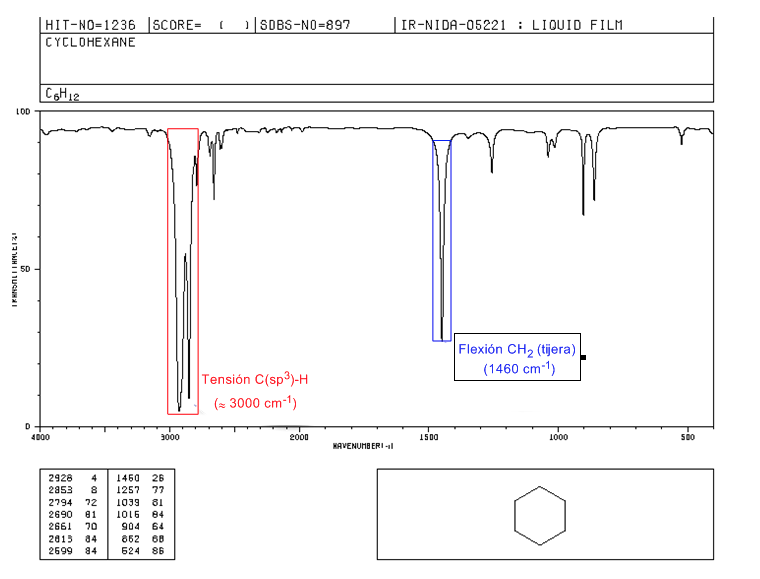
Si noti l'assenza della banda di flessione simmetrica metilica che gli alcani presentano a 1375 c m − 1 .
Spettro IR degli alcheni
• Tensione C(sp2)-H: 3100 -3000 cm-1
• Tensione C=C: 1600 cm-1
• Flessione fuori dal piano (oop) del legame C=CH: 1000 - 650 cm-1. Questo tipo di banda permette di conoscere il grado di sostituzione dell'alchene.
Spettro dell'1-pentene
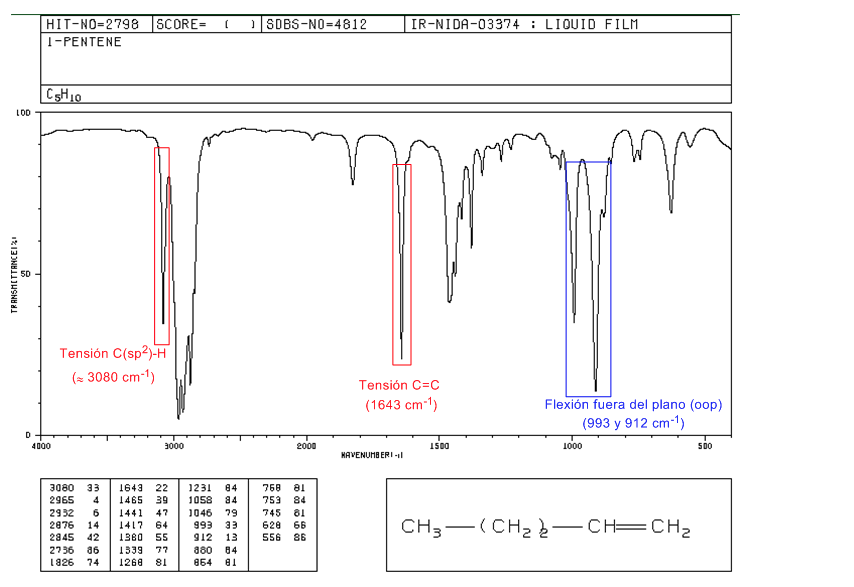
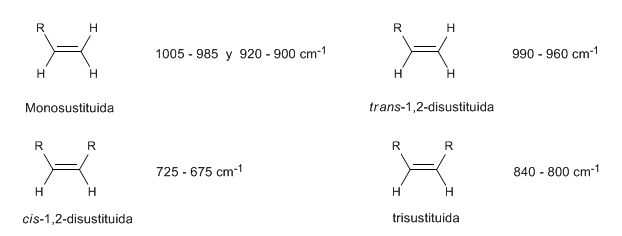
Negli alcheni monosostituiti, come l'1-pentene, le curve CH fuori dal piano producono due bande situate a 1005-985 e 920-900 c m − 1 .
Stereochimica e spettroscopia IR degli alcheni
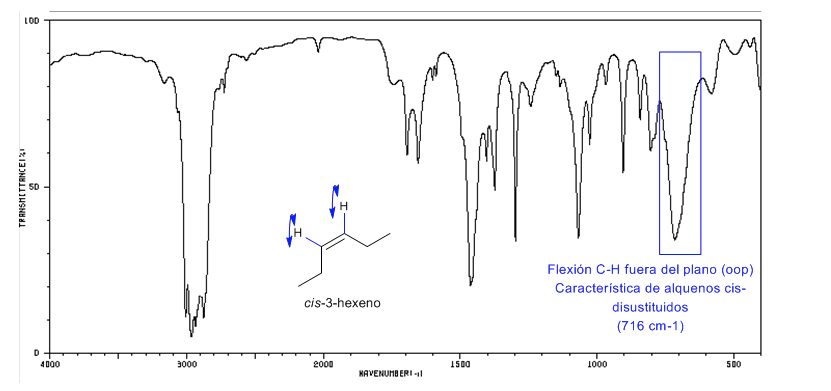
Gli alcheni cis-disostituiti presentano una banda di flessione CH fuori dal piano che consente di distinguerli. Questa banda appare tra 725-675 c m − 1
Spettro di Cis-3-esene
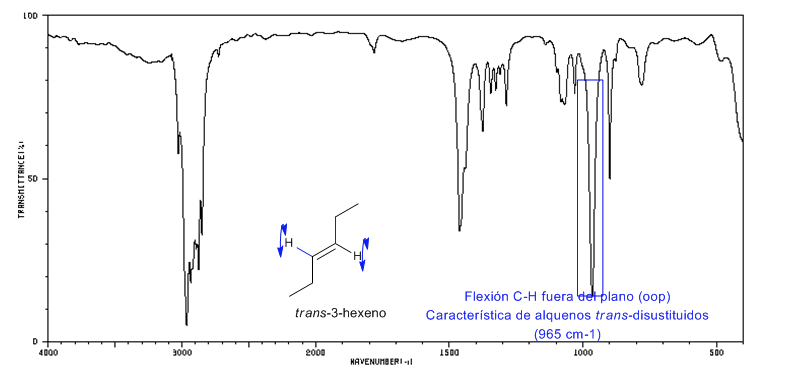
Spettro del Trans-3-esene
Gli alcheni trans-disostituiti presentano una forte banda di assorbimento tra 980-965 c m − 1 che ne permette l'identificazione. Si noti la totale assenza della banda di tensione C=C a 1600 c m − 1 a causa della mancanza di polarità.
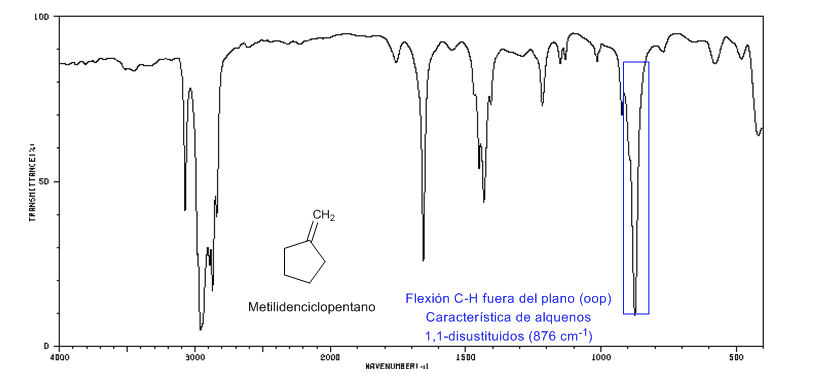
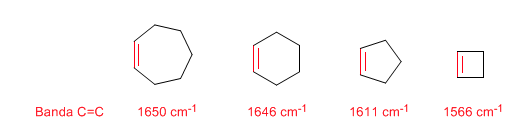
Ceppo dell'anello: legami endociclici C=C
Al diminuire delle dimensioni dell'anello, la banda di tensione dei legami C=C si sposta verso un numero minore di onde. L'eccezione del ciclopropene è attribuita all'accoppiamento tra le vibrazioni di tensione dei legami C=C e CC. Questo accoppiamento non si verifica nel ciclobutene perché i legami C=C e CC sono perpendicolari tra loro.
 Spettro IR del ciclopentene
Spettro IR del ciclopentene 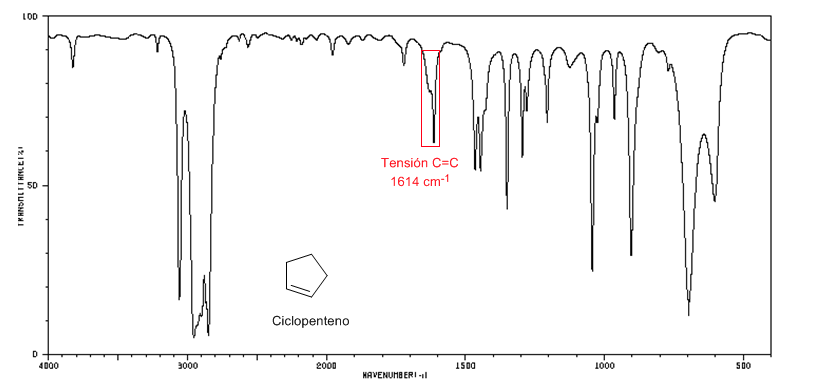
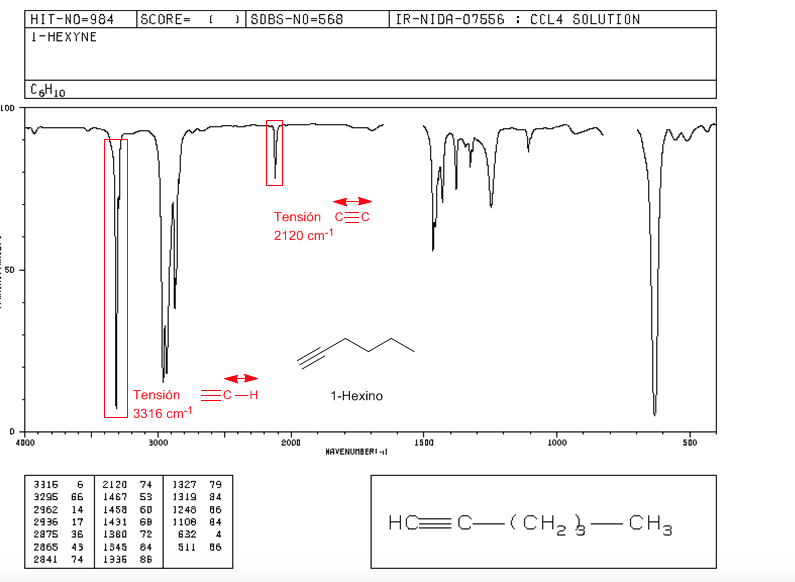
Tensione ≡ C − H : 3300 c m − 1
Tensione − C ≡ C − : 2150 c m − 1 . Gli alchini simmetrici non presentano questa banda, essendo molto deboli in quelle interne. La coniugazione abbassa leggermente il valore.
spettro di 1-esina
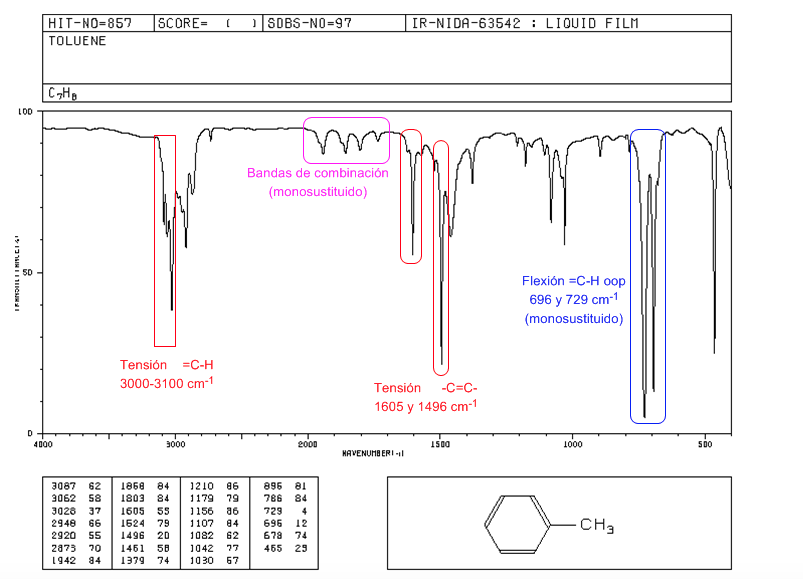
Spettro IR degli aromatici
Tensione =CH: 3100 cm-1
Tensione -DO=DO-: 1600 e 1475 cm-1
Flessione =CH fuori dal piano: 900-690 cm-1
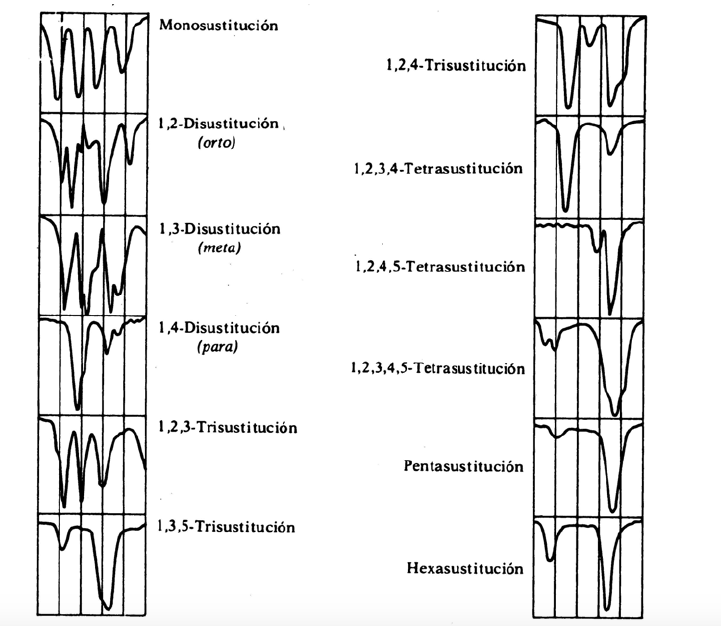
Le vibrazioni oop insieme agli armonici e alle bande di combinazione che compaiono tra 2000 e 1667 cm-1 ci permettono di conoscere il grado di sostituzione del benzene.
bande di ricambio
![]()
Spettri di alcoli e fenoli
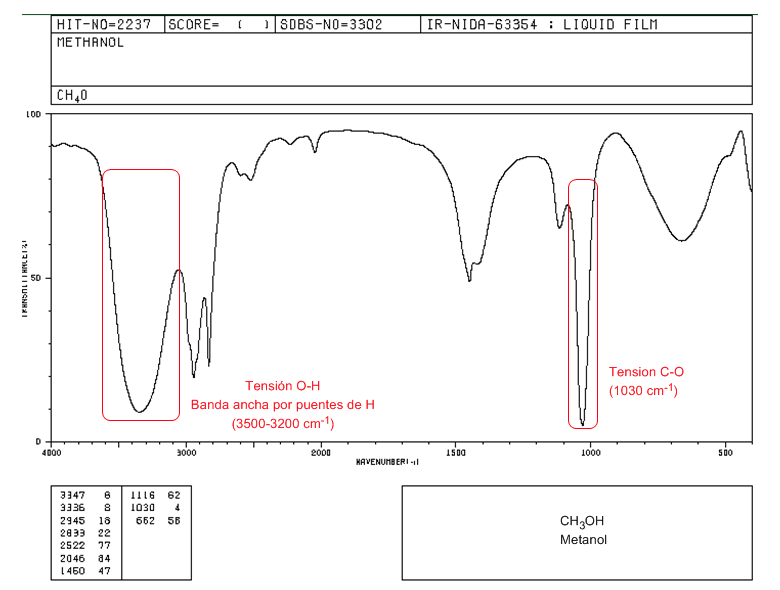
Tensione OH: Banda larga da 3500 a 3200 cm-1. In assenza di legame idrogeno, appare come un picco acuto a 3650-3600 cm-1.
Tensione CO: Banda tra 1250-1000 cm-1. Permette di distinguere tra alcoli primari (1050 cm-1), secondari (1100 cm-1), terziari (1150 cm-1) e fenoli (1220 cm-1).
Spettro IR di un alcol primario (metanolo)
Nello spettro del metanolo si osserva la banda di tensione OH molto ampia, dovuta alla formazione di legami idrogeno. La banda di tensione del CO esce con un numero d'onda basso (1030) perché è un alcol senza sostituenti.
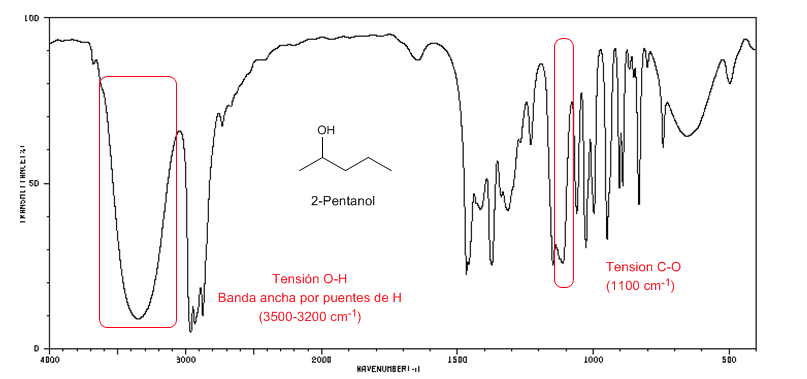
Spettro IR degli alcoli secondari (2-pentanolo)
Si osservi lo spostamento della banda CO verso un numero maggiore di onde rispetto al metanolo.
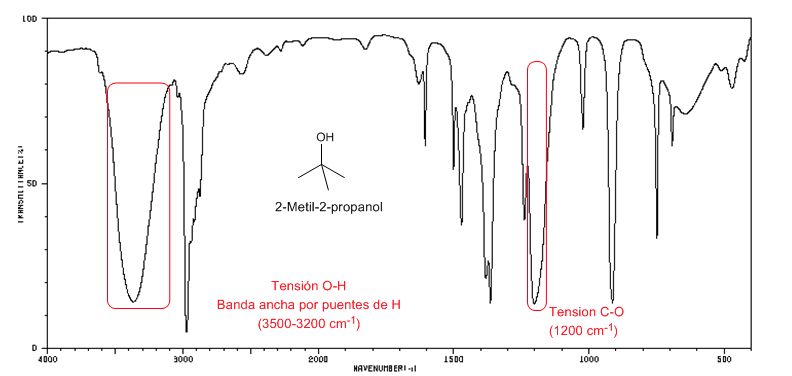
 Spettro IR degli alcoli terziari (2-metil-2-propanolo)
Spettro IR degli alcoli terziari (2-metil-2-propanolo)Gli alcoli terziari hanno la banda CO spostata a frequenze più alte rispetto agli alcoli primari e secondari.
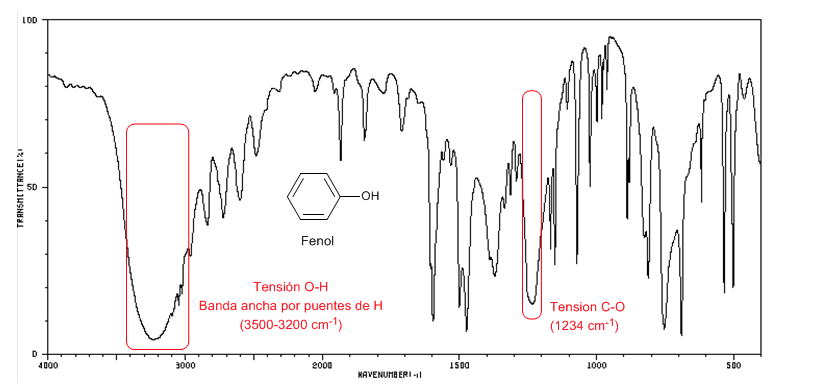
Il fenolo presenta una banda di assorbimento di CO superiore a 1200 cm-1
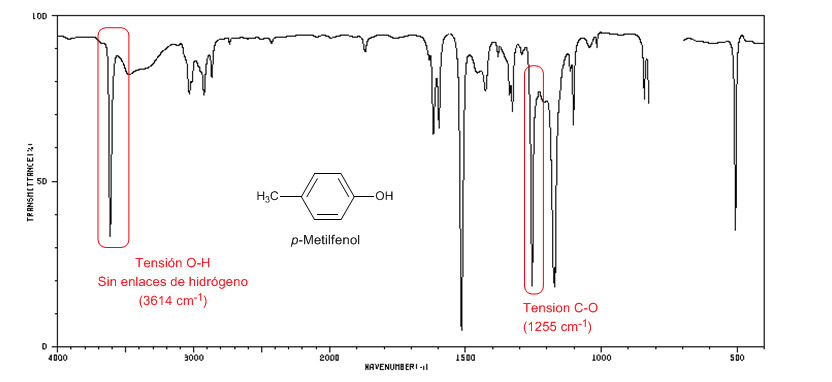
Il seguente spettro mostra la banda elastica OH in assenza di legame idrogeno.
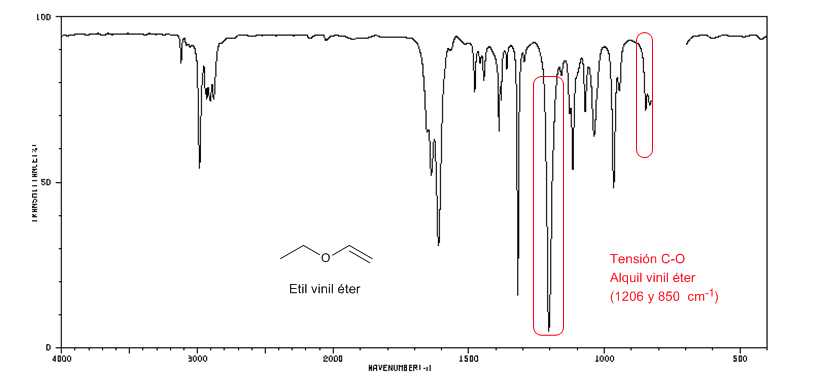
Gli alchil vinil eteri (CH2=CH-OR) presentano due bande a 1220 e 850 cm-1. Quest'ultimo molto debole.
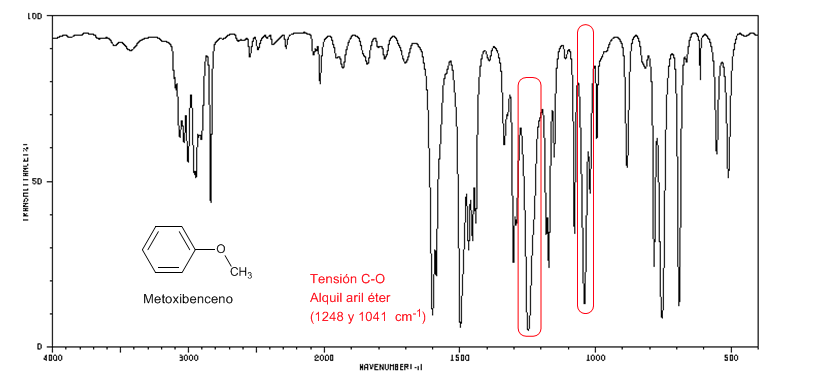
Gli aril alchil eteri (Ar-OR) presentano due bande a 1250 e 1040 cm-1
Spettro IR dell'1-metossiesano
L'etil vinil etere presenta due bande a 1220 e 850 cm-1, quest'ultima molto debole.
Il metossibenzene presenta due bande a 1250 e 1040 cm-1
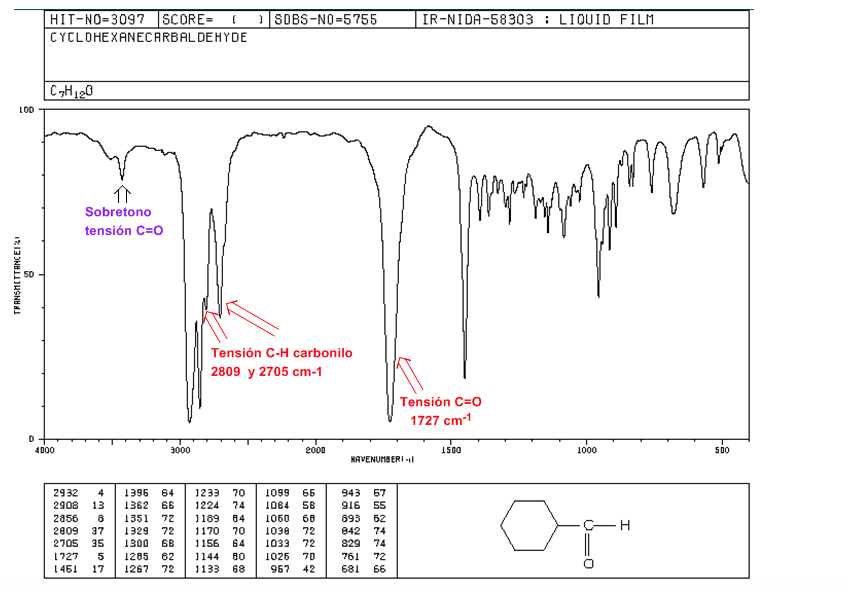
aldeidi
Tensione C=O: 1725 cm-1
Ceppo carbonilico CH: due bande deboli a 2850 e 2750 cm-1. La fascia a 2850 tende a sovrapporsi a quella di tensione C(sp3)-H
C=O Tono di tensione oltre 3500 cm-1.
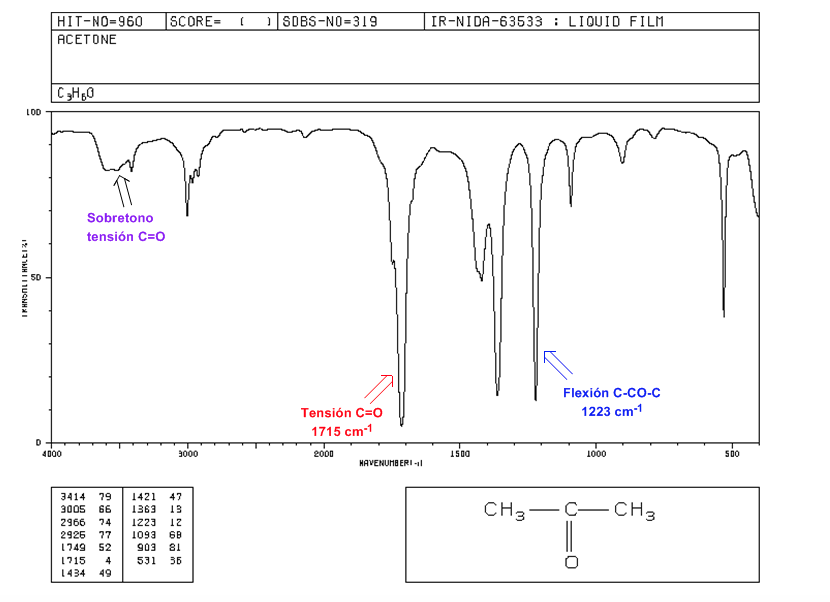
Tensione C=O: Banda intensa a 1715 cm-1.
Flessione C-CO-C: 1300 - 1100 cm-1.
Tono tensione C=O: da 3500 a 3350 cm-1.
Effetto della coniugazione sulla banda di tensione C=O
Spettro IR di acidi carbossilici e derivati
Acidi carbossilici
Tensione OH: Da 3400 a 2400 cm-1. Molto ampia a causa della formazione di legami a idrogeno.
Tensione C=O: 1730-1700 cm-1
Tensione CO: 1320-1200 cm-1
Flessione COH (oop): fascia a campana a 900 cm-1
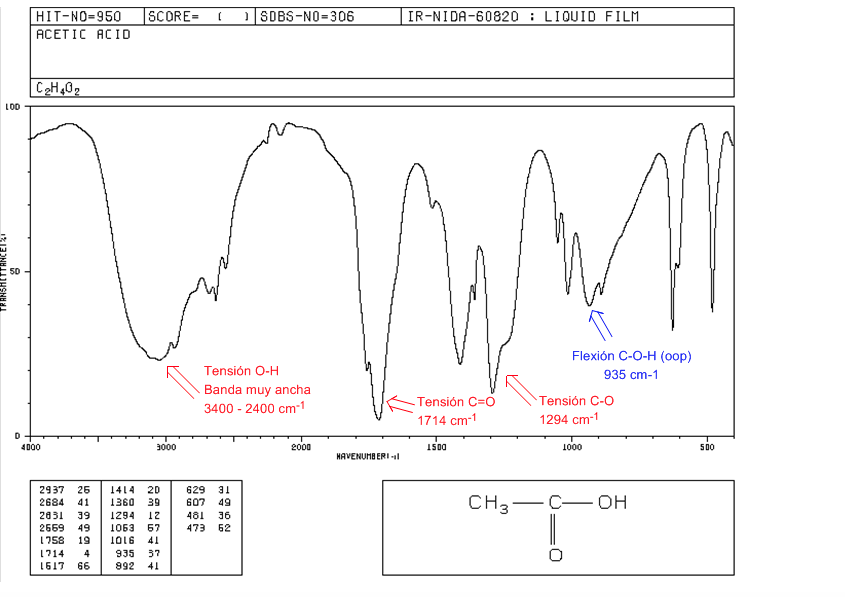
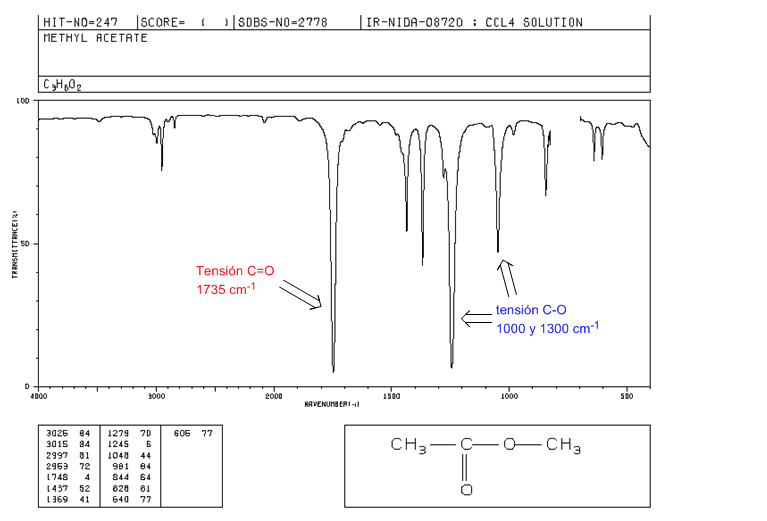
esteri
Tensione C=O a 1735 cm-1. Se sono presenti doppi legami coniugati con il carbonile, la banda si sposta a valori inferiori. Quando il doppio legame è sul gruppo alcossilico (-OR) dell'estere, si osserva uno spostamento verso valori più alti.
Tensione CO: 2 bande a 1300 e 1000 cm-1. Essendo più ampio e più intenso quello osservato a 1300.
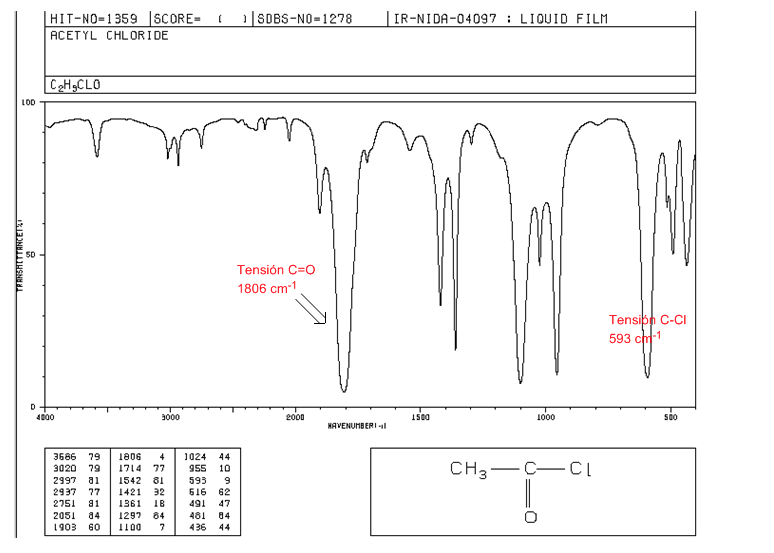
alogenuri acidi
Tensione C=O: 1810 - 1775 cm-1
Tensione C-Cl: banda intensa 730 - 550 cm-1
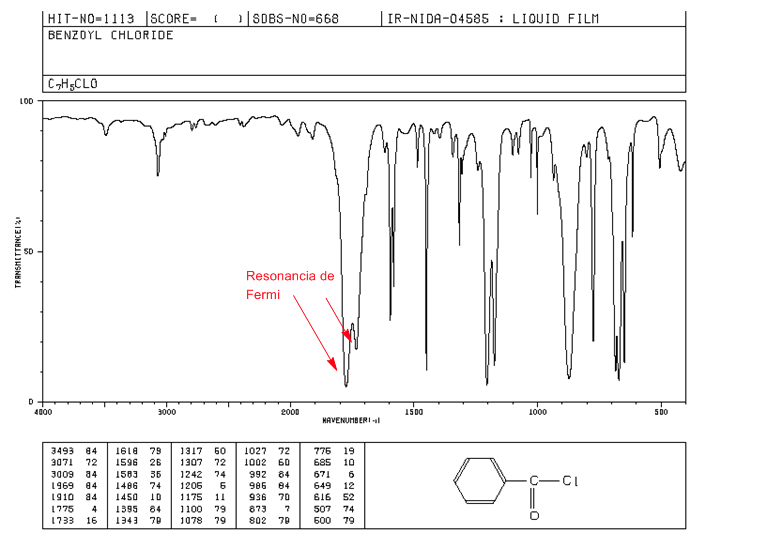
Risonanza di Fermi
Gli armonici sono transizioni vibrazionali dallo stato fondamentale a stati eccitati superiori. Le frequenze di assorbimento sono la frequenza dell'assorbimento fondamentale. La risonanza di Fermi risulta dall'accoppiamento di una banda fondamentale di assorbimento con una banda armonica o combinata.
Gli alogenuri alcanoilici aromatici mostrano due bande di deformazione C=O mediante risonanza di Fermi.
 Nitrili
Nitrili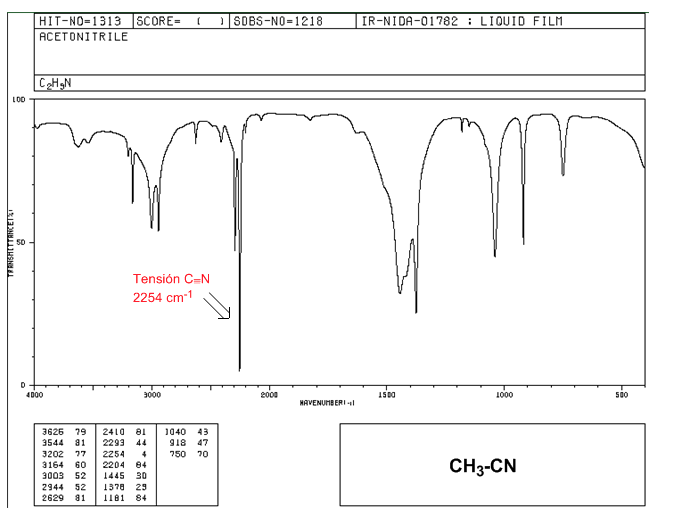
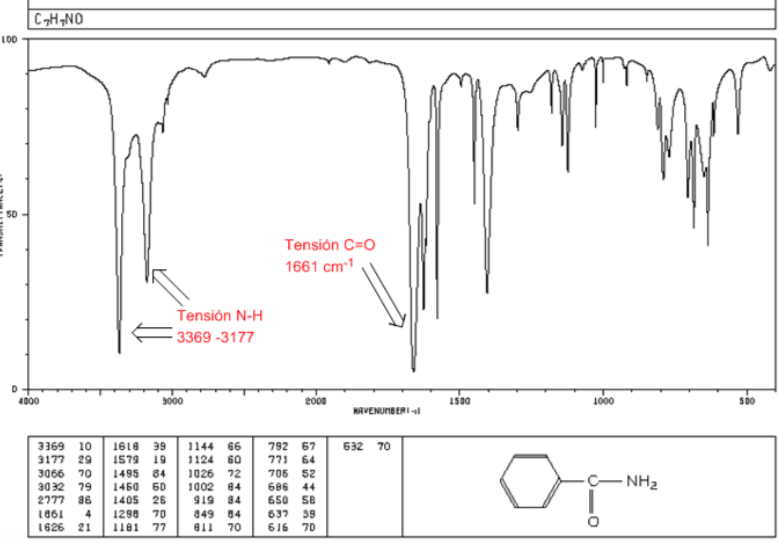
ammidi
Tensione C=O: 1680 - 1630 cm-1
Tensione NH: Tra 3350 e 3180 cm-1. Le ammidi primarie hanno due bande, mentre le ammidi secondarie hanno solo una banda.
Flessione NH: 1640 - 1550 cm-1
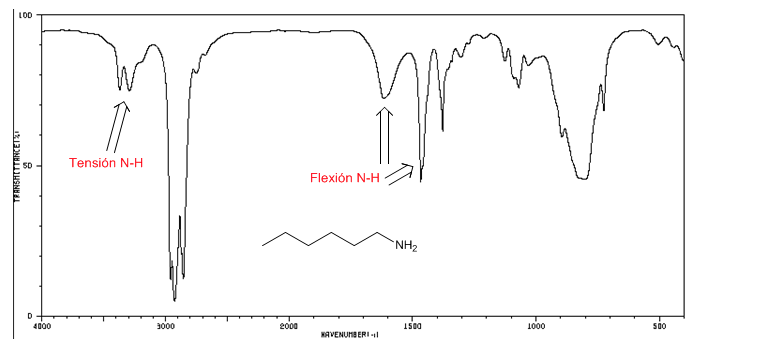
Spettro IR delle ammine
Tensione NH: tra 3500 e 3300 cm-1. Le ammine primarie presentano due bande (simmetrica e asimmetrica), quelle secondarie una sola banda.
Flessione NH: ammine primarie due bande a 1640 e 1560 cm-1. Secondaria una fascia a 1500 cm-1
Spettro IR dell'1-esano-ammina
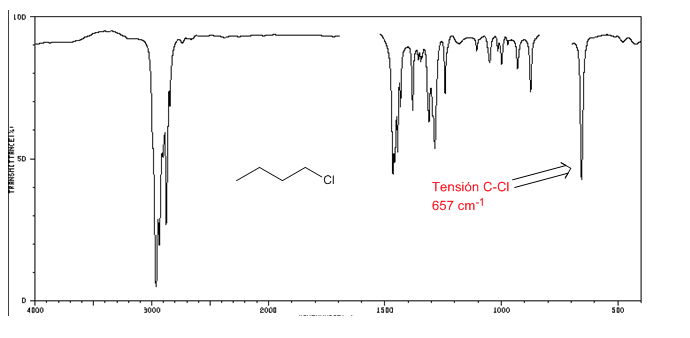
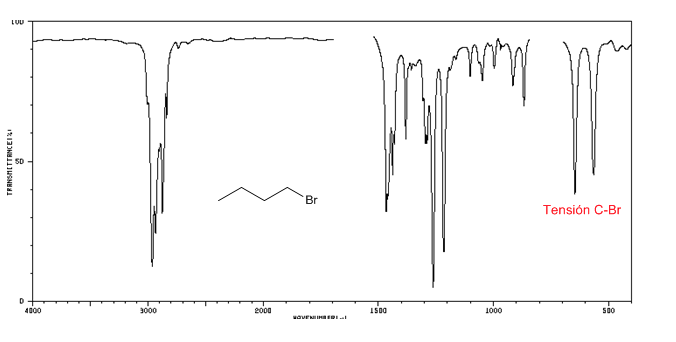
Spettro IR degli alogenuri alchilici
Tensione C-Cl: 785 - 540 cm-1
Ension C-Br: 650 - 510 cm-1
 Fonti consigliate per ampliare la ricerca:
Fonti consigliate per ampliare la ricerca:
1. Spettroscopia di emissione Aga Fano SA. (Consultato il 10 settembre 2007). http://hiq.aga.com.co/International/Web/LG/CO/likelgspgco.nsf/DocByAlias/anal_icp .
2. Alonso, P. et al. chimica Cou..Ed. McGraw Hill. 1990.
3. Álvarez Jiménez, MD e Gómez del Río, MI Guida Didattica Chimica Analitica II. UNED. 1999.
4. Arrivi Jimeno Sirò; Burriel Barceló Fernando; Hernandez Mendez Gesù; Lucena Conte Felipe. Chimica analitica qualitativa. ISBN: 8497321405. ISB. 2006.
5. Ayres, Gilbert H. Analisi chimica quantitativa. Edizioni del Castello, 4a ed . ISBN: 8421902806. 1981.
6. Bermejo Barrera. M. del Pilar. Chimica analitica generale, quantitativa e strumentale. Editoriale Paraninfo. 7a edizione. ISBN: 8428318093. 1990.
7. Blanco, M., Cerdá, V. e Sanz Medel, A., Analytical Atomic Spectroscopy, Pubblicazioni dell'Università Autonoma di Barcellona. 1990.
8. Brodo. RW, spettroscopia chimica, New York 1952.
9. Burriel, MF, Lucena, CF. Chimica analitica quantitativa. Edizione rivoluzionaria. L'Avana.1978.
10. Burriel, F. Chimica analitica qualitativa. Editoriale Paraninfo. ISBN: 8497321405. pp 1072. , 2003.