spectroscopie infrarouge
La spectrométrie infrarouge est extrêmement utile pour les déterminations qualitatives de composés organiques et pour déduire des structures moléculaires à partir de leurs groupes fonctionnels de composés organiques et inorganiques.
Dans l'analyse qualitative, la spectroscopie infrarouge peut être utilisée pour l'identification de substances pures ou pour l'absorption, la localisation et l'identification d'impuretés.
Pour localiser une impureté dans une substance, une comparaison est faite entre le spectre de la substance étudiée et un échantillon de la substance pure. Les impuretés provoquent l'apparition de bandes d'absorption supplémentaires dans le spectre.
Dans l'IR, ils trouvent également une utilisation croissante dans l'analyse quantitative, le principal domaine d'application de ce type d'analyse étant la quantification des polluants atmosphériques issus des procédés industriels.
Une partie du spectre électromagnétique qui s'étend de 0,8 à 1000μm (ce qui correspond au nombre d'onde entre 12800 et 10 cm-1), est considérée comme la région infrarouge qui se divise en trois régions appelées :
a).- IR proche. b).- IR fondamental ou moyen c).- IR lointain
Chaque type de liaison absorbe le rayonnement infrarouge à une fréquence différente, ce qui permet de déterminer le type de groupes fonctionnels de la molécule étudiée. Les spectrophotomètres infrarouges fonctionnent dans l'infrarouge moyen et balayent de 4000 c m − 1 à 400 c m − 1
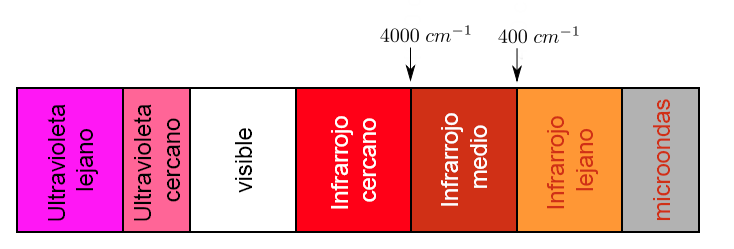
Cependant, la région d'importance analytique est la région IR fondamentale puisque la plupart des instruments infrarouges couvrent cette région.
La plupart des matériaux organiques et inorganiques présentent une absorption et le spectre est principalement causé par l'étirement et la flexion vibratoires au sein de la molécule. Le spectre infrarouge est l'une des propriétés les plus caractéristiques d'un composé puisqu'il n'y a pas deux spectres identiques pour deux composés différents, c'est comme une empreinte digitale.
Dans la région IR fondamentale, il y a deux régions, l'une d'elles est la soi-disant
les groupes fonctionnels de 4000 cm-1 à 1300 cm-1, et la région des doigts de 1300 cm-1 à 670 cm-1.
Au niveau des groupes fonctionnels, la position du pic d'absorption est plus ou moins élevée en fonction uniquement du groupe fonctionnel où arrive l'absorption et non de la structure moléculaire complète. La position des pics dans la région du doigt dépend de la structure moléculaire complète.
Caractéristiques que doit avoir une vibration pour produire une bande d'absorption :
Le rayonnement incident doit avoir une fréquence égale à la fréquence de la vibration qu'il va produire.
Que la vibration résultante produit un changement dans le moment dipolaire, c'est-à-dire que la vibration n'absorbera pas le rayonnement infrarouge, s'il n'y a pas de changement dans le moment dipolaire, cela s'appelle une vibration inactive et ils seront actifs lorsqu'il y aura ledit changement dans le dipôle moment.
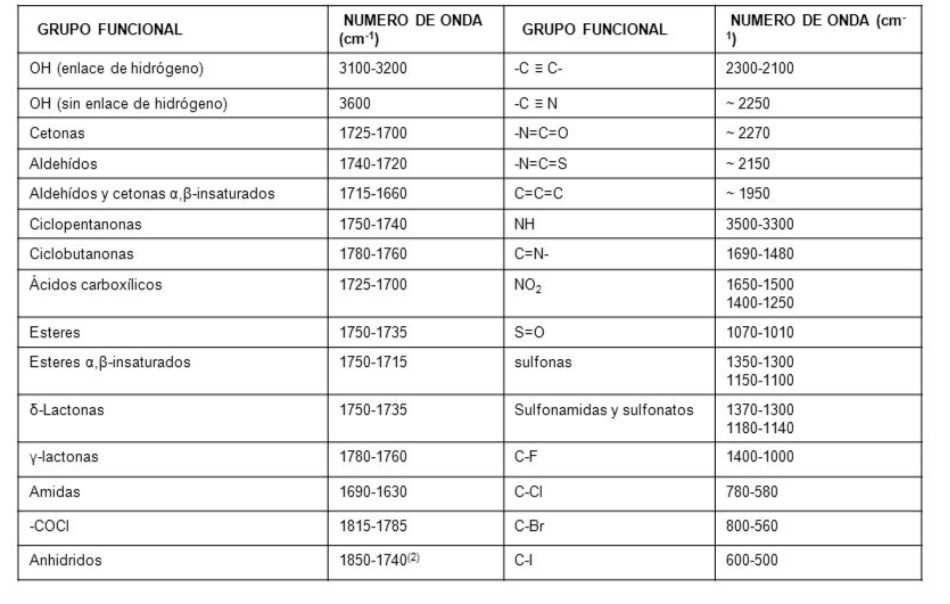
Traditionnellement, l'axe des abscisses des spectres infrarouges utilise le nombre d'ondes ( ν ¯ , lire "nu bar"') et est défini comme l'inverse de la longueur d'onde en cm. ν ¯ = 1 λ . L'axe des ordonnées représente le pourcentage de rayonnement transmis (transmittance) qui est représenté par % T . La forme du spectre infrarouge de l'hexane est illustrée ci-dessous.
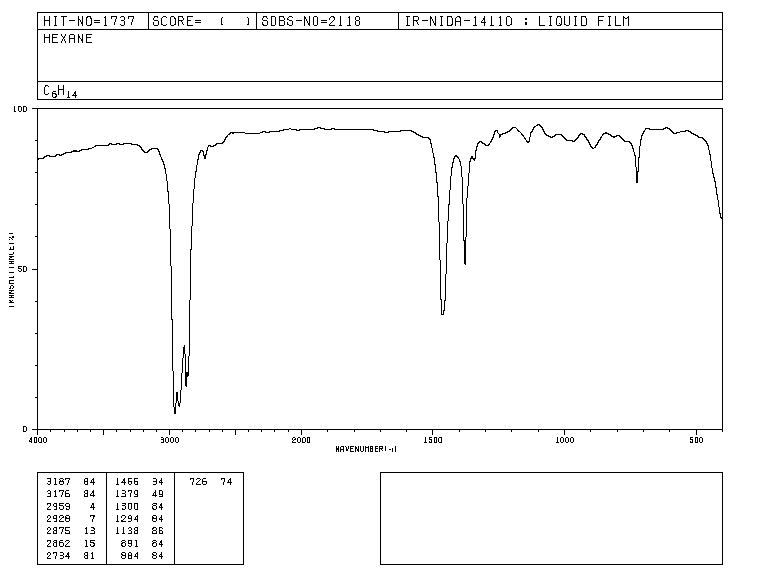
Les bandes représentent les zones où les liaisons de la molécule absorbent le rayonnement infrarouge. Dans les bandes, la transmittance est petite et l'absorbance est grande.
types de vibrations
Vibration de tension (étirement). Les atomes maintenus ensemble par des liaisons simples, doubles ou triples se rapprochent et s'éloignent dans la direction de la liaison, tout comme deux masses attachées à un ressort oscillent.
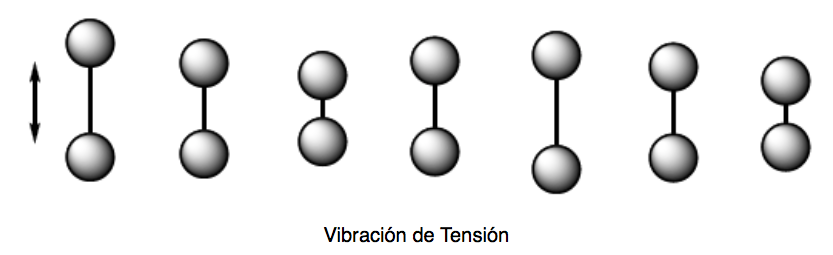
Il existe deux modes de vibration de tension : symétrique et asymétrique.
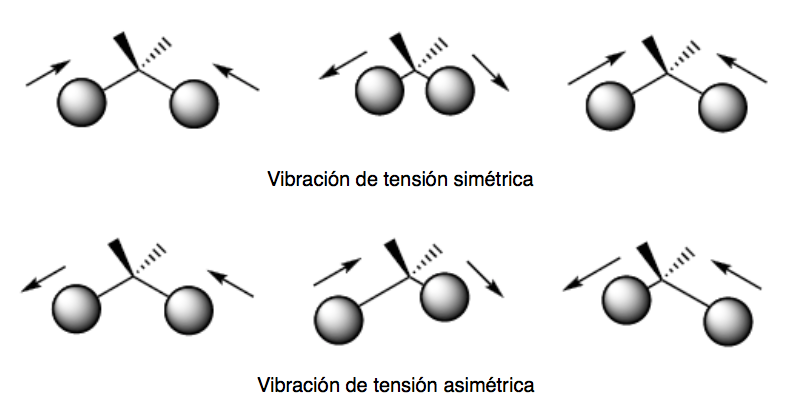
vibrations de flexion. Les atomes vibrent de sorte que les angles varient, mais pas les longueurs de liaison. Il existe quatre modes de vibrations de flexion : cisaillement, basculement, remue-ménage et torsion.
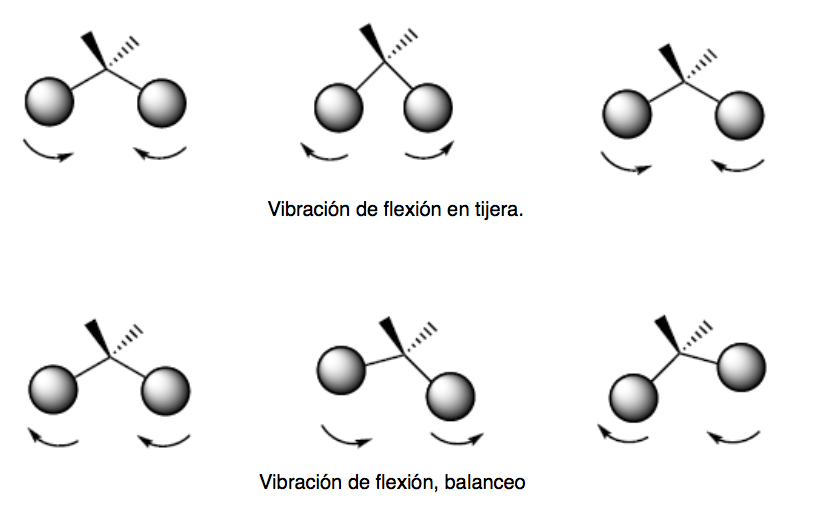
Ces deux modes de vibration ont lieu dans le plan qui contient les trois atomes qui participent à la vibration.
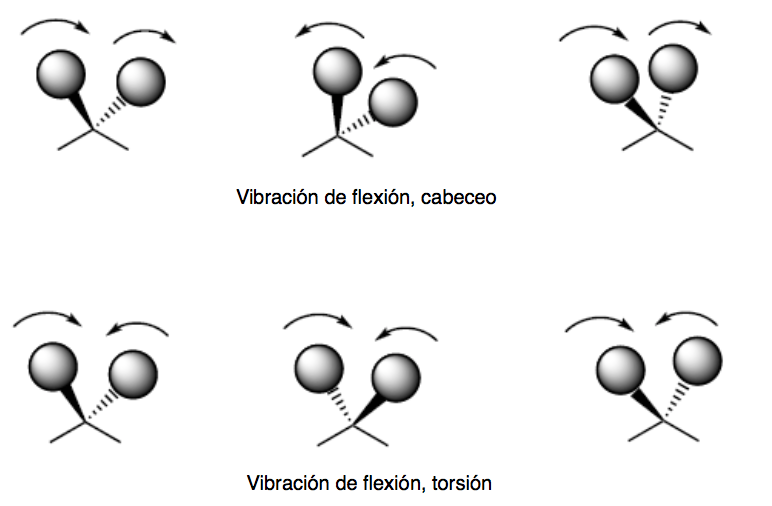
Les modes de vibration de tangage et de torsion se produisent hors du plan (Out of plane) et sont généralement représentés par Oop.
oscillateur harmonique quantique
Les vibrations moléculaires peuvent être étudiées avec le modèle d'oscillateur harmonique quantique. L'énergie est donnée par :
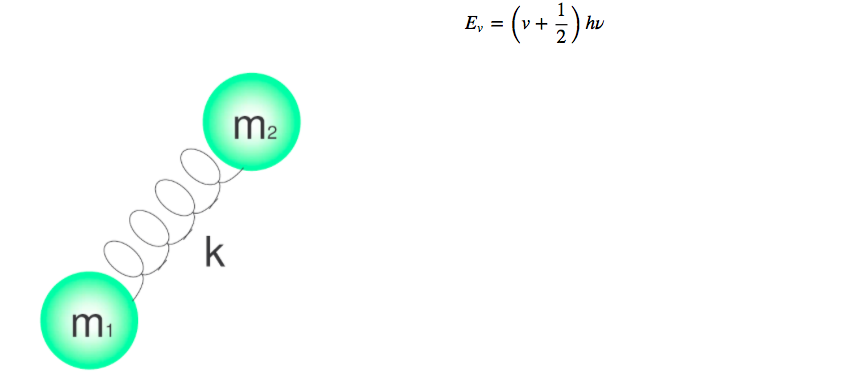
Les différents niveaux d'énergie sont donnés par le nombre quantique v, qui prend les valeurs 0.1.2.3.4.....
h est la constante de Planck et ν est la fréquence de l'oscillateur qui est donnée par l'expression :
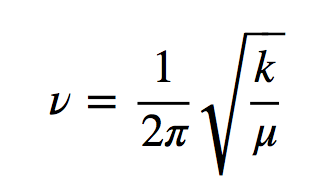
où k est la constante de force du ressort et μ la masse réduite du système
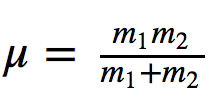
En divisant la fréquence par la vitesse de la lumière, on obtient le nombre d'ondes ν ¯
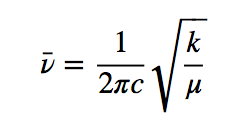
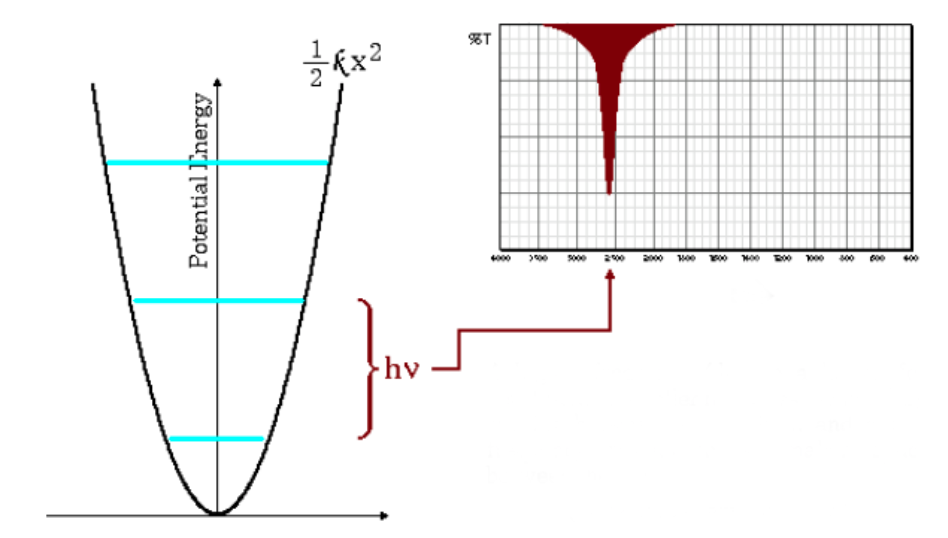
L'équation ci-dessus indique que de petites masses réduites (atomes de faible masse) et des constantes de force élevées (liaisons fortes) conduisent à des fréquences élevées. Dans ces conditions, les bandes d'absorption émergent à des nombres d'onde élevés.
Comme on peut le voir sur le graphique, les hautes fréquences donnent lieu à un plus grand espacement entre les niveaux d'énergie.
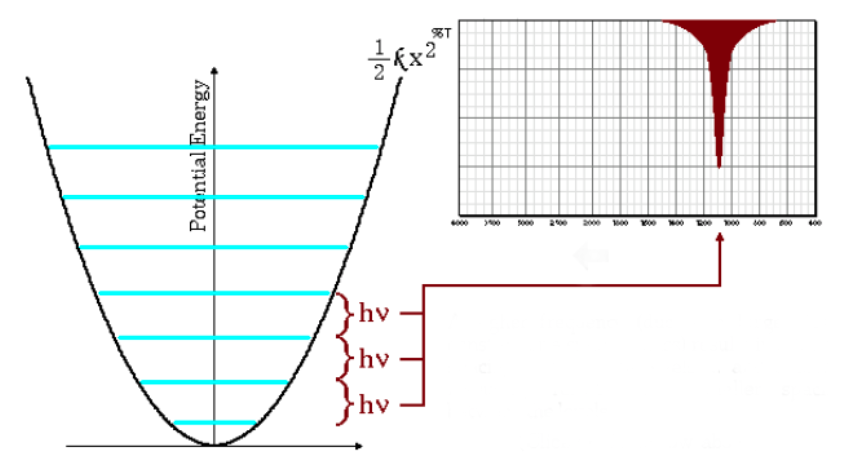
Basses fréquences d'absorption
L'équation ci-dessus aussi indique que de grandes masses réduites et de petites constantes de force (liaisons faibles) conduisent à des basses fréquences. Dans ces conditions, les bandes d'absorption sortent à de faibles nombres d'onde.
Comme on peut le voir sur le graphique, les basses fréquences donnent lieu à moins d'espacement entre les niveaux d'énergie.
Principales vibrations moléculaires
Spectre IR
Dans un spectre infrarouge, la fréquence (en nombre d'onde) est tracée en fonction du pourcentage de lumière transmise (transmittance). Le pourcentage de transmission est défini comme le quotient entre l'intensité de la lumière transmise à travers l'échantillon, I M , et l'intensité de la lumière du faisceau de référence I R multiplié par 100.
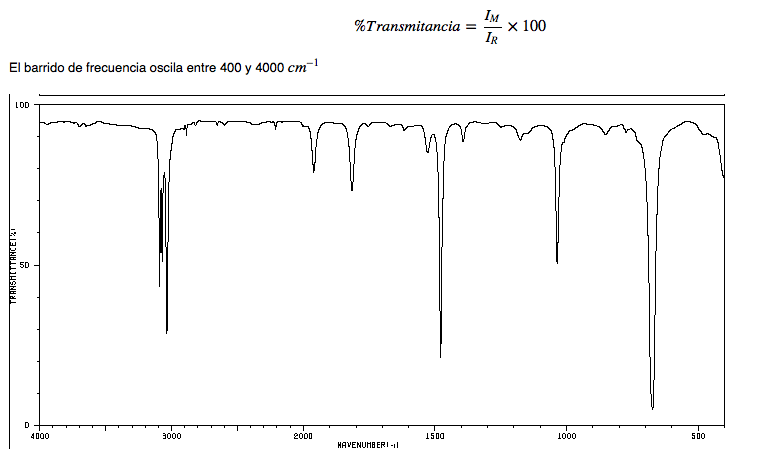
Spectre IR des alcanes
• Déformation CH : les alcanes présentent des vibrations de déformation CH légèrement inférieures à 3000 c m − 1
• CH flexion : les CH 2 de la chaîne présentent des vibrations de flexion (ciseaux) à 1465 c m − 1 , tandis que les méthyles produisent une bande à 1375 c m − 1 due à la vibration de flexion symétrique et une autre à 1450 c m − 1 due à vibration de flexion asymétrique. Toutes les bandes de pompes sont d'intensité moyenne.
Notez que la bande de flexion asymétrique du méthyle chevauche la bande de flexion en ciseaux du CH 2 .
Spectre IR de l'hexane
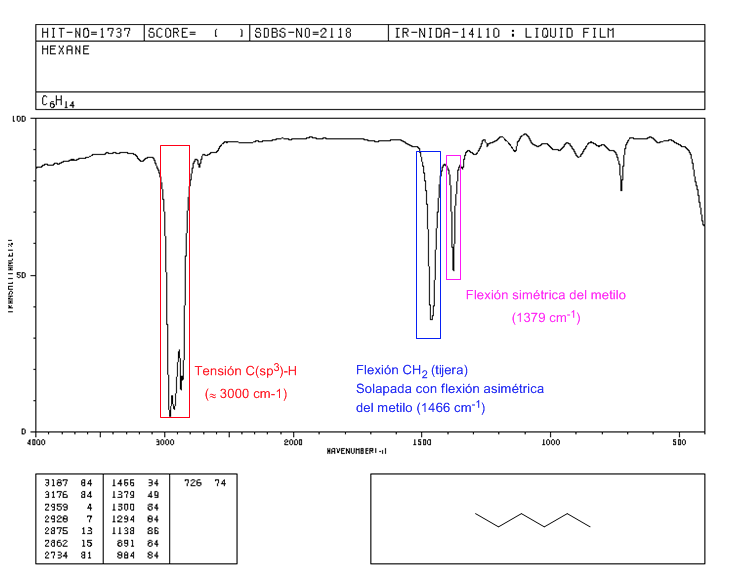
Spectre du 2,2-diméthylbutane
La présence du groupement tert-butyle produit le dédoublement de la bande de flexion symétrique en deux bandes à 1390 et 1370 c m - 1 . La bande à 1390 est moitié moins forte que celle à 1370.
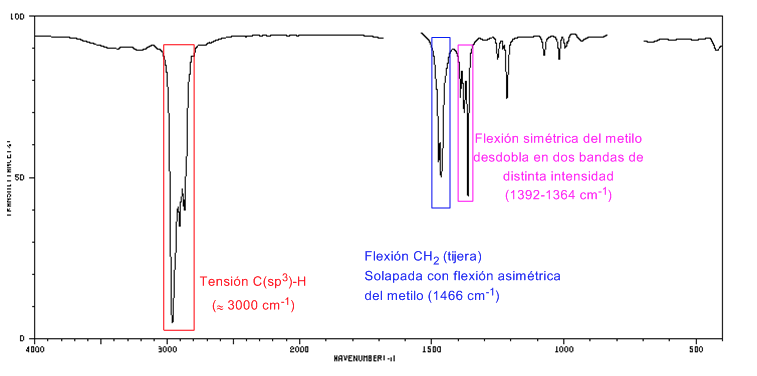
Spectre IR des cycloalcanes
Les cycloalcanes ont un spectre IR très similaire aux alcanes avec une bande d'étirement CH légèrement inférieure à 3000 c m − 1 et une bande de flexion CH en ciseaux pour CH 2 à 1465 c m − 1 . La principale différence avec les alcanes est l'absence de la bande d'étirement symétrique du méthyle. 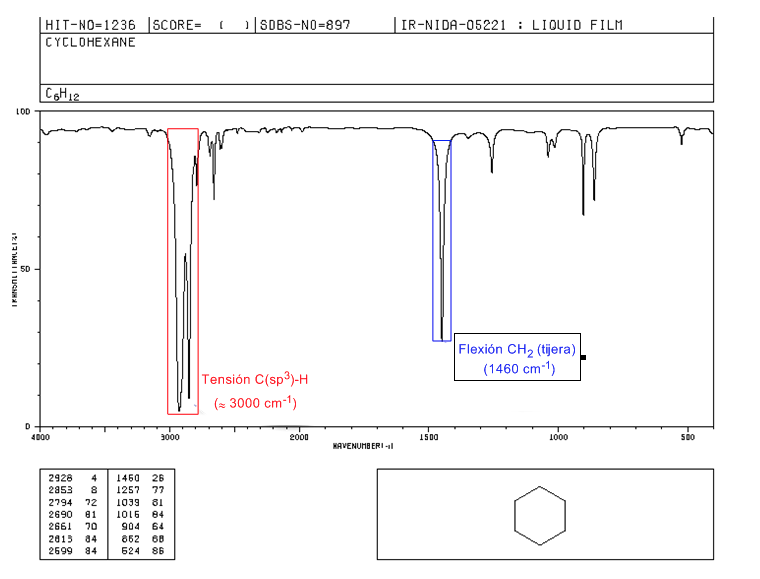
Notez l'absence de la bande de flexion symétrique méthyle que les alcanes présentent à 1375 c m − 1 .
Spectre IR des alcènes
• Tension C(sp2)-H : 3100 -3000 cm-1
• Tension C=C : 1600 cm-1
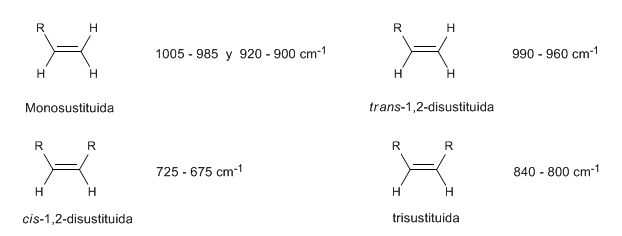
• Flexion hors plan (oop) de la liaison C=CH : 1000 - 650 cm-1. Ce type de bande permet de connaître le degré de substitution de l'alcène.
Spectre de 1-pentène
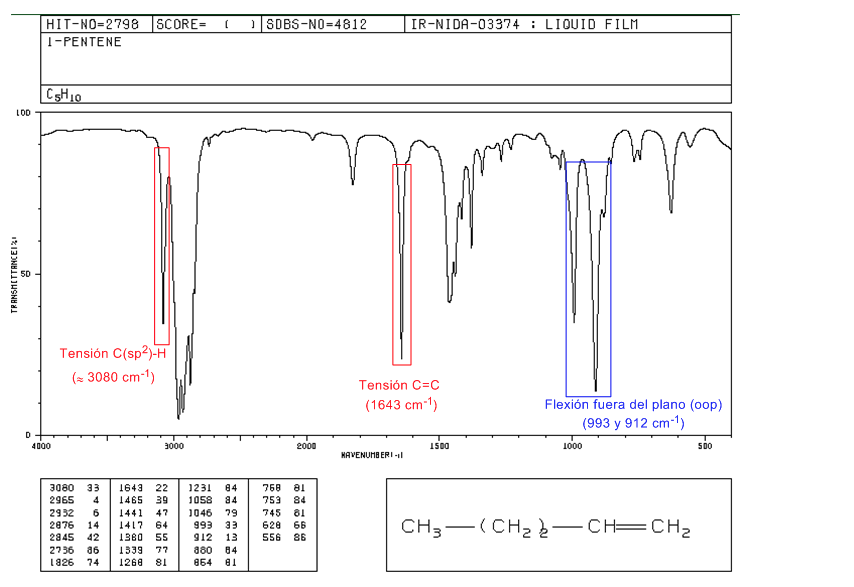
Dans les alcènes monosubstitués, tels que le 1-pentène, les coudes CH hors du plan produisent deux bandes situées à 1005-985 et 920-900 c m - 1 .
Stéréochimie et spectroscopie IR des alcènes
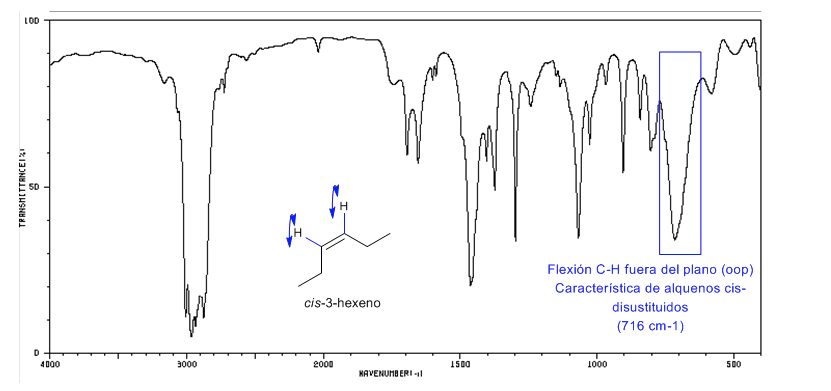
Les alcènes cis-disubstitués présentent une bande de flexion CH hors du plan qui permet de les distinguer. Cette bande apparaît entre 725-675 c m − 1
Spectre de Cis-3-hexène
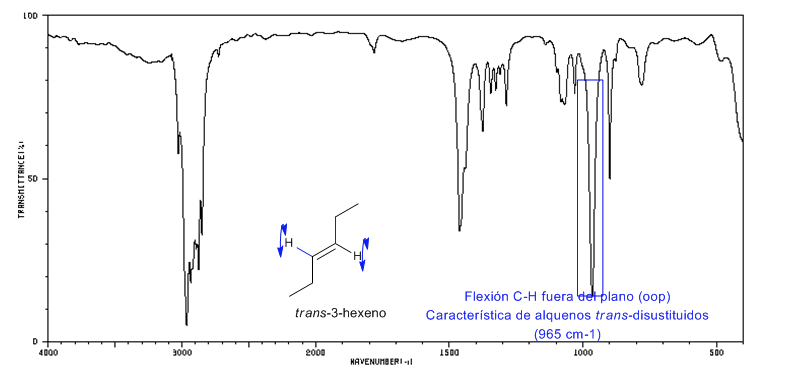
Spectre de Trans-3-hexène
Les alcènes trans-disubstitués présentent une forte bande d'absorption entre 980-965 c m − 1 qui permet leur identification. Notez l'absence totale de la bande de tension C=C à 1600 c m − 1 en raison du manque de polarité.
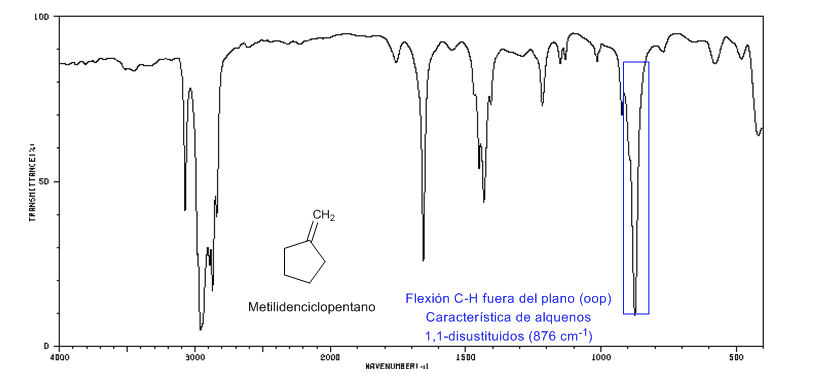
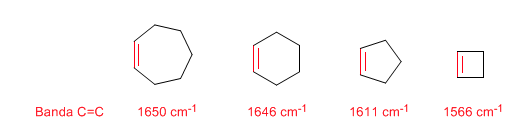
Souche annulaire : liaisons C = C endocycliques
Lorsque la taille de l'anneau diminue, la bande de tension des liaisons C=C se déplace vers un plus petit nombre d'ondes. L'exception du cyclopropène est attribuée au couplage entre les vibrations de tension des liaisons C=C et CC. Ce couplage ne se produit pas dans le cyclobutène car les liaisons C=C et CC sont perpendiculaires l'une à l'autre.
 Spectre IR du cyclopentène
Spectre IR du cyclopentène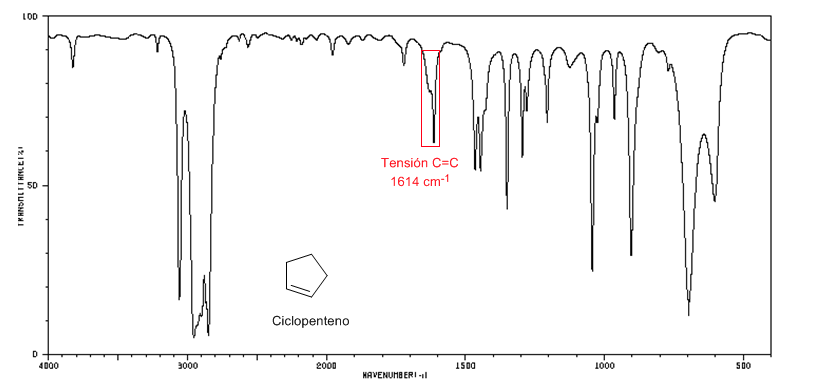
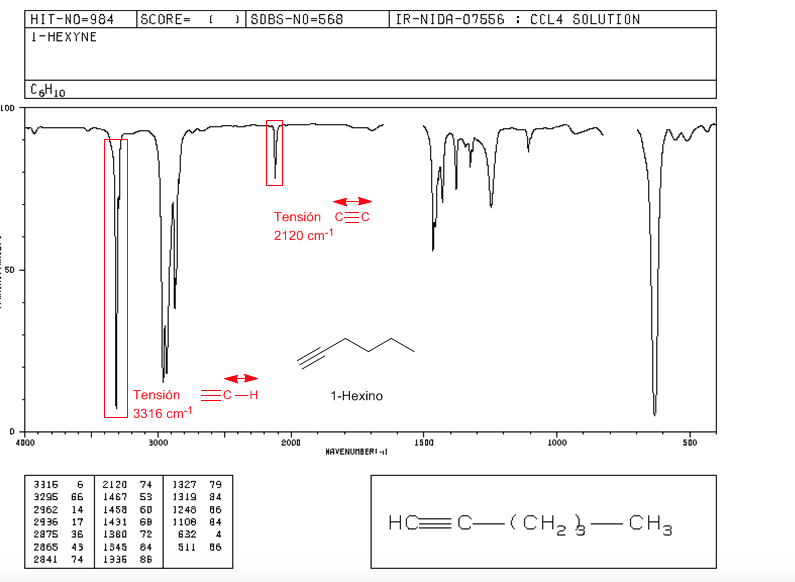
Traction ≡ C − H : 3300 c m − 1
Traction − C ≡ C − : 2150 c m − 1 . Les alcynes symétriques ne présentent pas cette bande, étant très faibles dans les internes. La conjugaison diminue légèrement la valeur.
spectre de 1-hexine
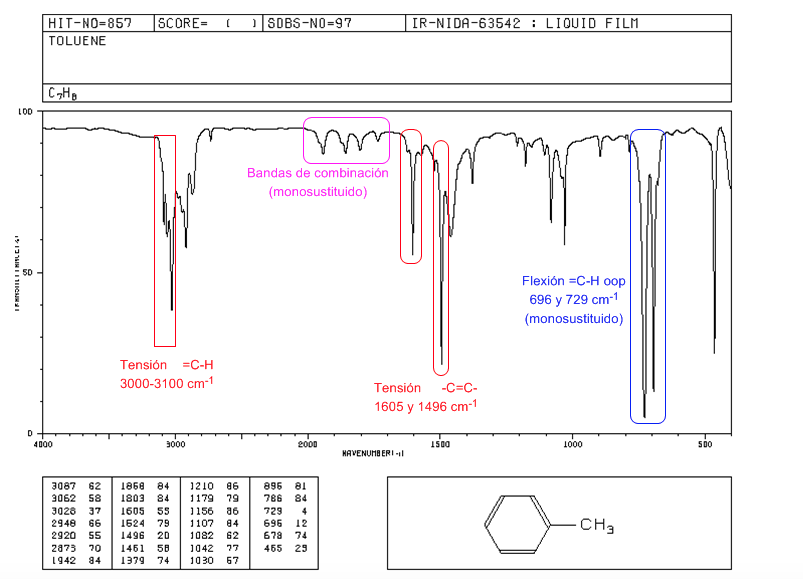
Spectre IR des aromatiques
Tension =CH : 3100 cm-1
Tension -C=C- : 1600 et 1475 cm-1
Flexion =CH hors plan : 900-690 cm-1
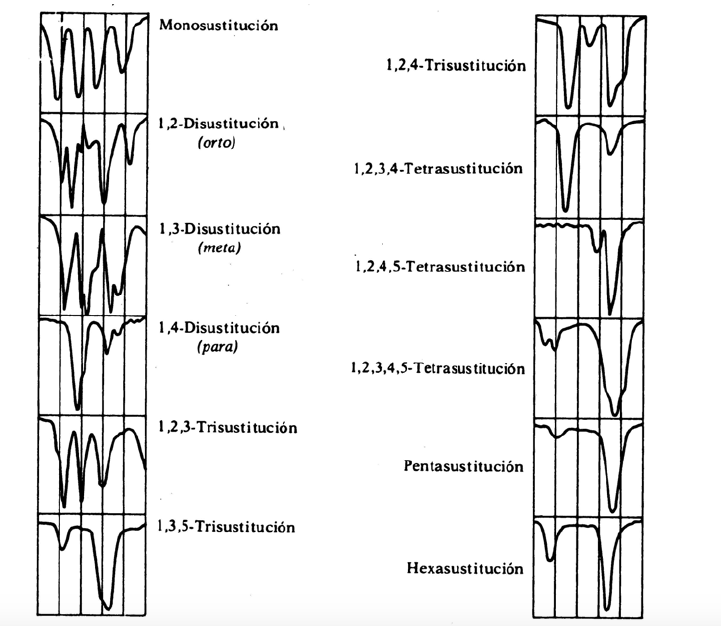
Les vibrations oop ainsi que les harmoniques et les bandes de combinaison qui apparaissent entre 2000 et 1667 cm-1 nous permettent de connaître le degré de substitution du benzène.
bandes de remplacement
![]()
Spectres d'alcools et de phénols
Tension OH : Bande large de 3500 à 3200 cm-1. En l'absence de liaison hydrogène, il apparaît comme un pic net à 3650-3600 cm-1.
Tension CO : Bande entre 1250-1000 cm-1. Il permet de distinguer les alcools primaires (1050 cm-1), secondaires (1100 cm-1), tertiaires (1150 cm-1) et phénols (1220 cm-1).
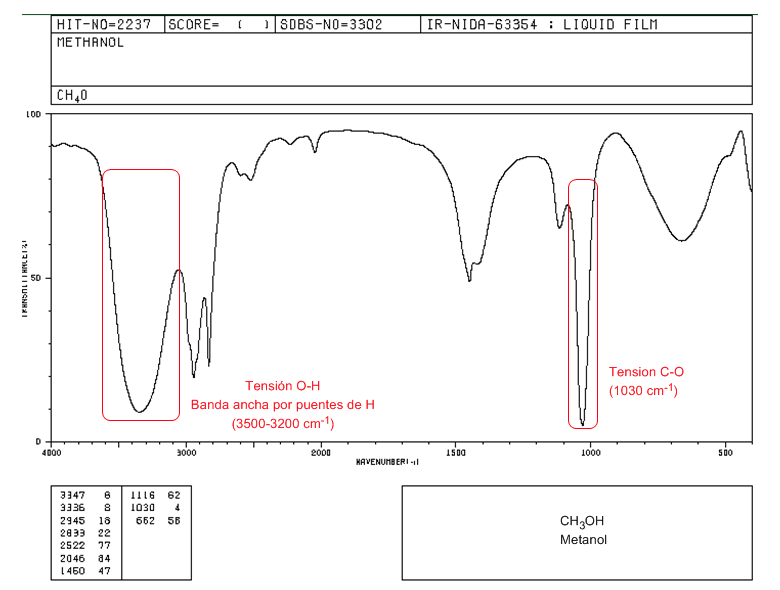
Spectre IR d'un alcool primaire (méthanol)
Dans le spectre du méthanol, on peut observer la très large bande de tension OH, due à la formation de liaisons hydrogène. La bande de tension CO sort à un nombre d'onde faible (1030) car c'est un alcool sans substituants.
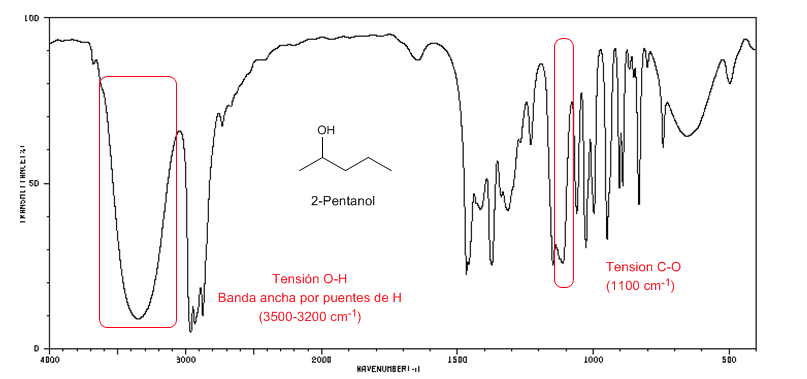
Spectre IR des alcools secondaires (2-pentanol)
Observez le déplacement de la bande CO vers un plus grand nombre d'ondes par rapport au méthanol.
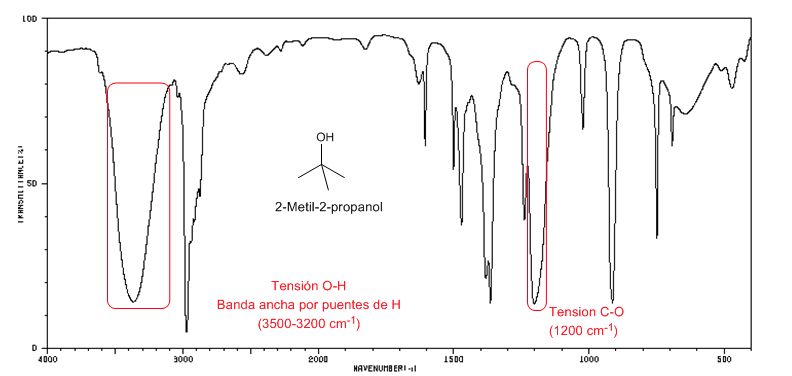
 Spectre IR des alcools tertiaires ( 2-méthyl-2-propanol)
Spectre IR des alcools tertiaires ( 2-méthyl-2-propanol)Les alcools tertiaires ont la bande CO décalée vers des fréquences plus élevées que les alcools primaires et secondaires.
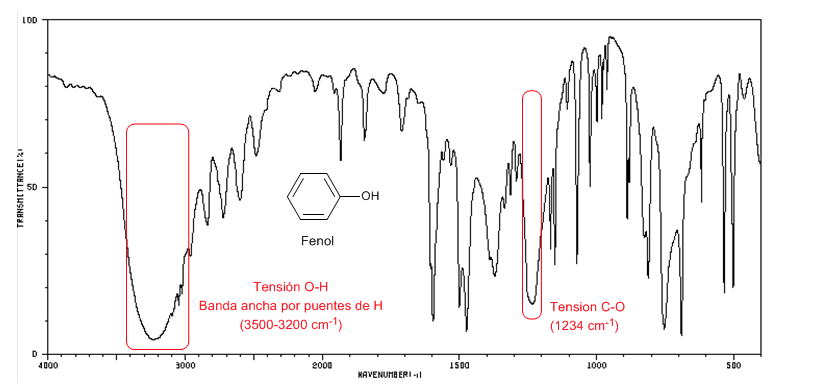
Le phénol présente une bande d'absorption du CO supérieure à 1200 cm-1
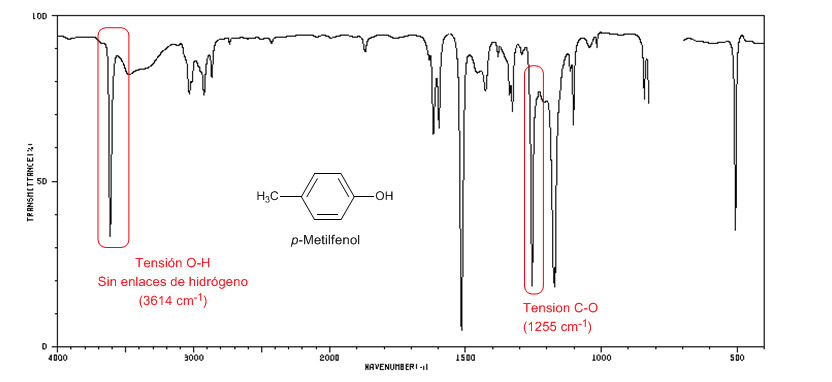
Le spectre suivant montre la bande d'étirement OH en l'absence de liaison hydrogène.
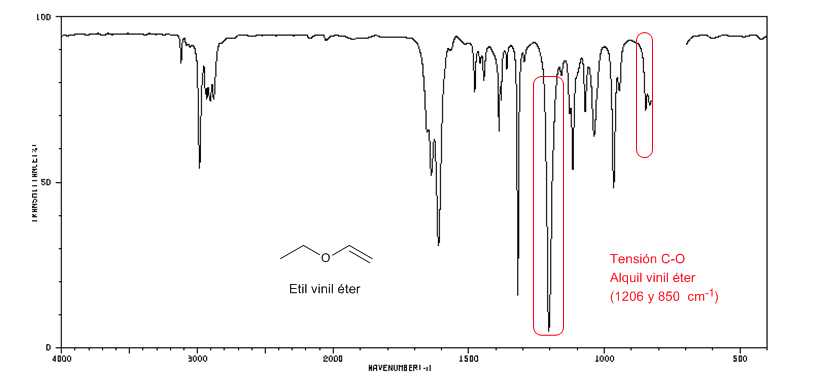
Les éthers alkyl vinyliques (CH2=CH-OR) présentent deux bandes à 1220 et 850 cm-1. Ce dernier très faible.
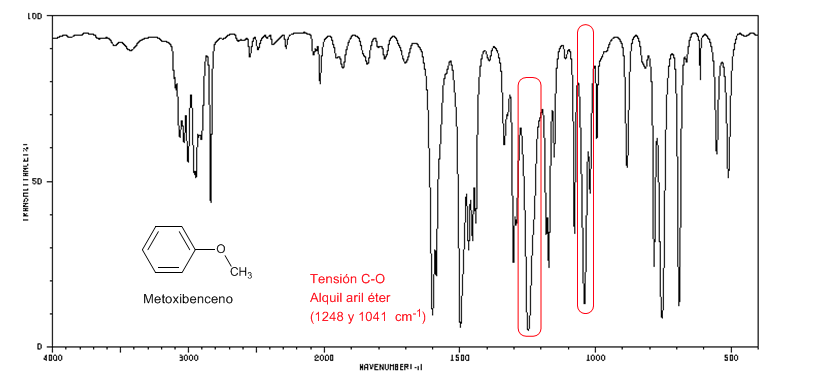
Les aryl alkyl éthers (Ar-OR) présentent deux bandes à 1250 et 1040 cm-1
Spectre IR du 1-méthoxyhexane
L'éther éthylvinylique présente deux bandes à 1220 et 850 cm-1, cette dernière est très faible.
Le méthoxybenzène présente deux bandes à 1250 et 1040 cm-1
aldéhydes
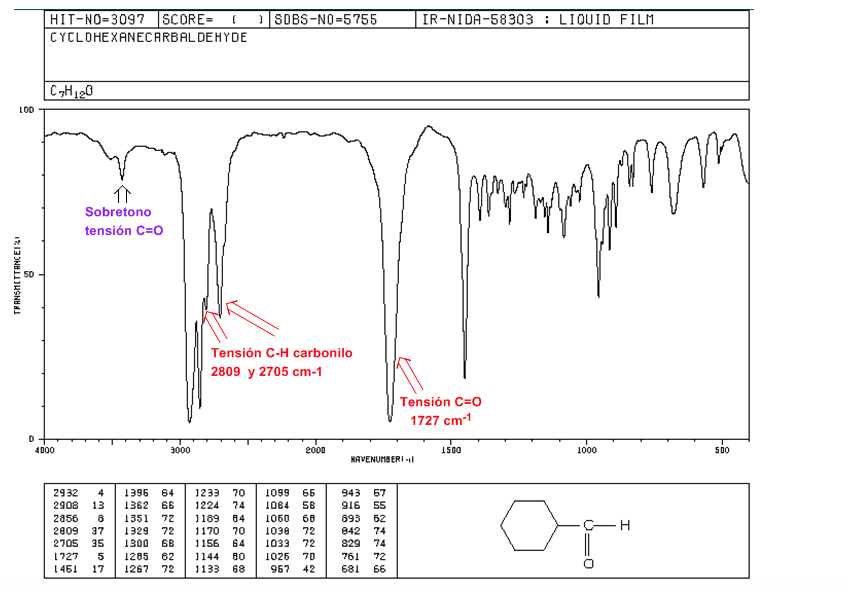
Tension C=O : 1725 cm-1
Souche carbonyle CH : deux bandes faibles à 2850 et 2750 cm-1. La bande à 2850 a tendance à se superposer à celle de la tension C(sp3)-H
C=O Tension Harmonique supérieure à 3500 cm-1.
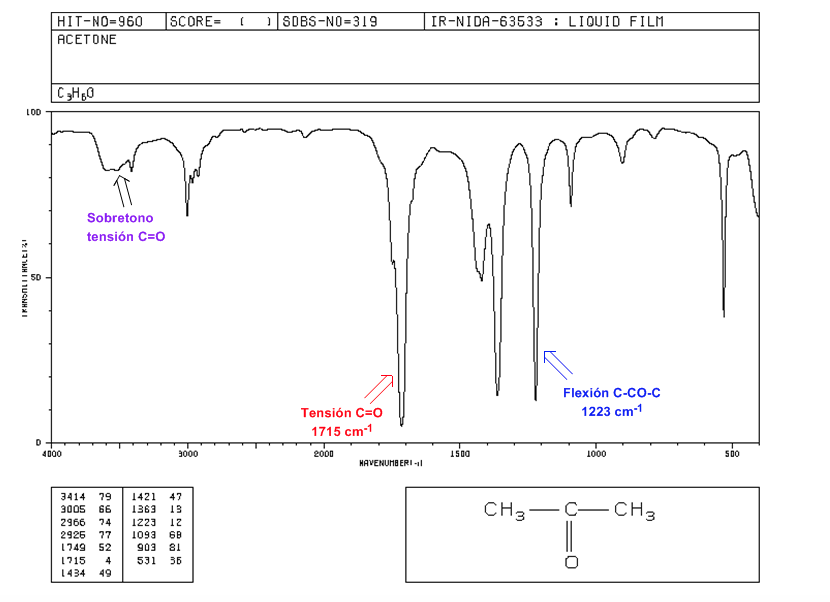
Tension C=O : Bande intense à 1715 cm-1.
Flexion C-CO-C : 1300 - 1100 cm-1.
Harmonique de tension C=O : de 3500 à 3350 cm-1.
Effet de la conjugaison sur la bande de tension C=O
Spectre IR des acides carboxyliques et dérivés
Acides carboxyliques
Tension OH : De 3400 à 2400 cm-1. Très large en raison de la formation de liaisons hydrogène.
Tension C=O : 1730-1700 cm-1
Tension CO : 1320-1200 cm-1
COH Flexion (oop) : Bande en cloche à 900 cm-1
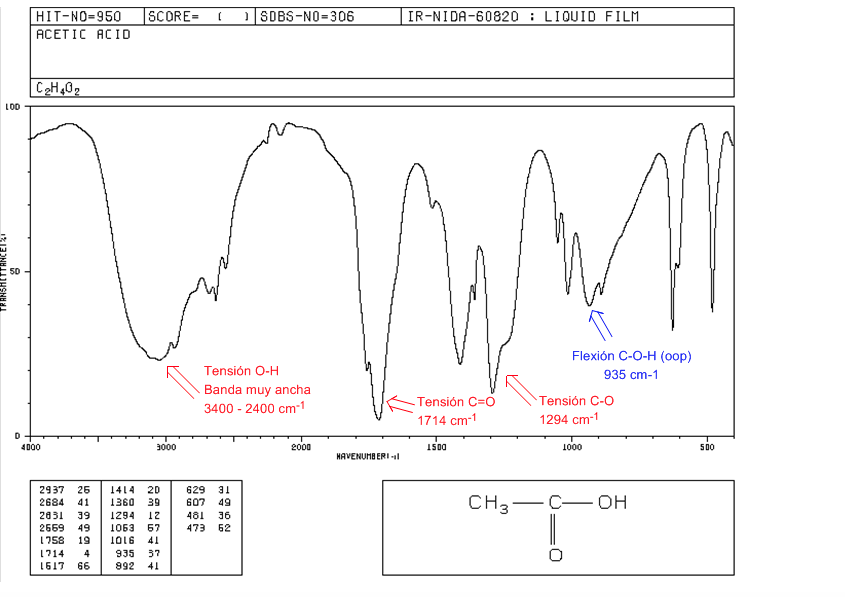
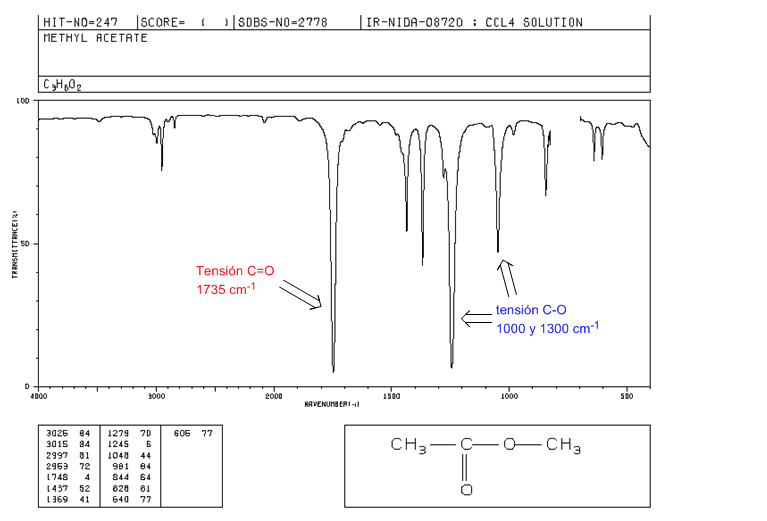
esters
Tension C=O à 1735 cm-1. S'il existe des doubles liaisons conjuguées avec le carbonyle, la bande se déplace vers des valeurs inférieures. Lorsque la double liaison est sur le groupe alcoxy (-OR) de l'ester, on observe un déplacement vers des valeurs plus élevées.
Tension CO : 2 bandes à 1300 et 1000 cm-1. Étant plus large et plus intense celle observée à 1300.
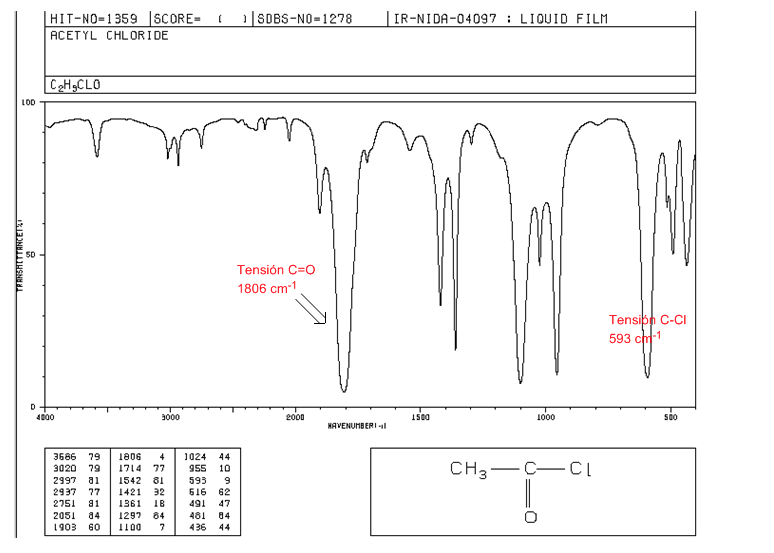
halogénures d'acide
Tension C=O : 1810 - 1775 cm-1
Tension C-Cl : bande intense 730 - 550 cm-1
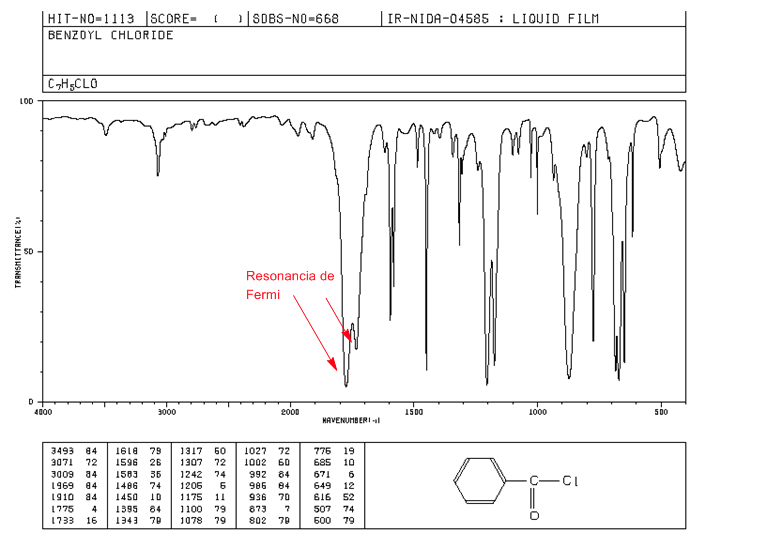
Résonance de Fermi
Les harmoniques sont des transitions vibrationnelles de l'état fondamental vers des états excités plus élevés. Les fréquences d'absorption sont la fréquence de l'absorption fondamentale. La résonance de Fermi , résulte du couplage d'une bande d'absorption fondamentale avec une harmonique ou une bande de combinaison.
Les halogénures d'alcanoyle aromatiques présentent deux bandes de déformation C = O par résonance de Fermi.
 Nitriles
Nitriles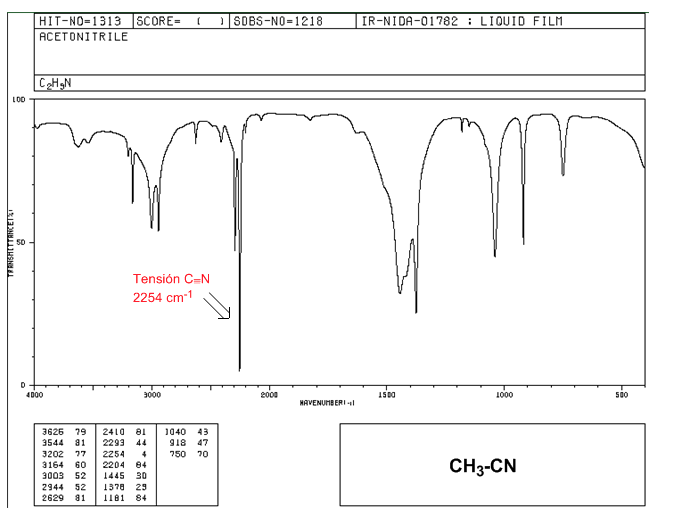
amides
Tension C=O : 1680 - 1630 cm-1
Tension NH : Entre 3350 et 3180 cm-1. Les amides primaires ont deux bandes, tandis que les amides secondaires n'ont qu'une seule bande.
Flexion NH : 1640 - 1550 cm-1
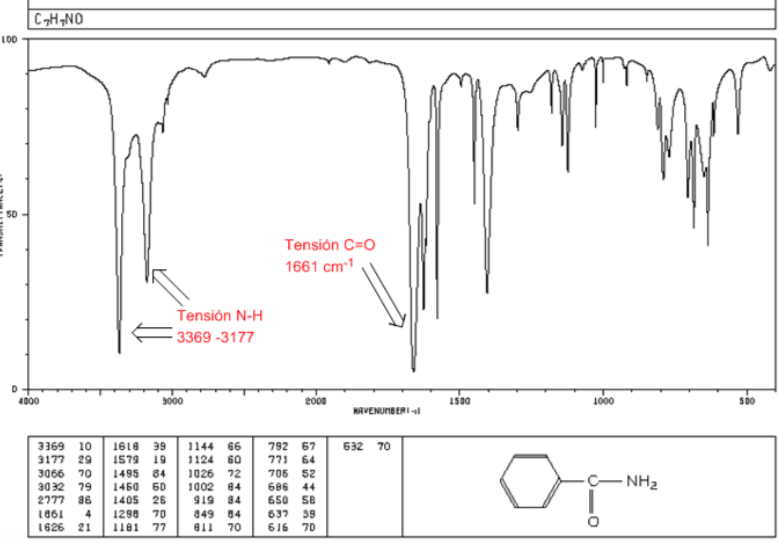
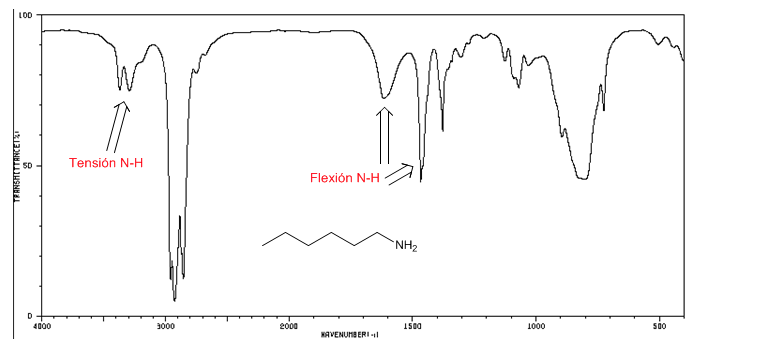
Spectre IR des amines
Tension NH : entre 3500 et 3300 cm-1. Les amines primaires présentent deux bandes (symétrique et asymétrique), les secondaires une seule bande.
Flexion NH : Amines primaires deux bandes à 1640 et 1560 cm-1. Secondaires une bande à 1500 cm-1
Spectre IR de la 1-hexane-amine
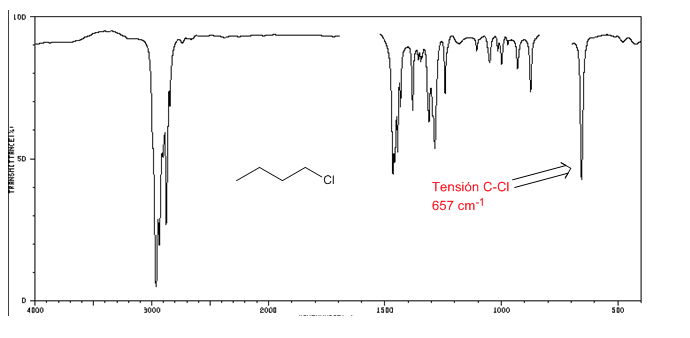
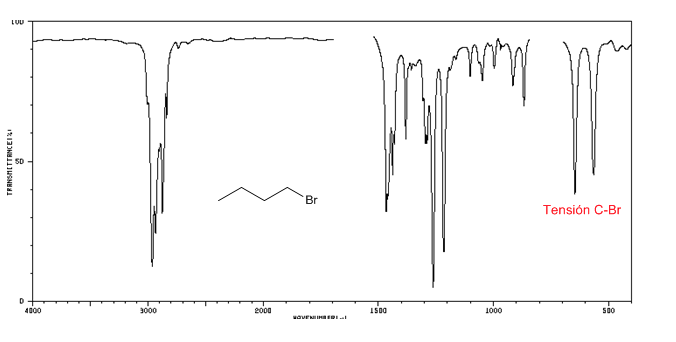
Spectre IR des halogénures d'alkyle
Tension C-Cl : 785 - 540 cm-1
Ension C-Br : 650 - 510 cm-1
 Sources recommandées pour élargir la recherche :
Sources recommandées pour élargir la recherche :
1. Spectroscopie d'émission Aga Fano SA. (Consulté le 10 septembre 2007). http://hiq.aga.com.co/International/Web/LG/CO/likelgspgco.nsf/DocByAlias/anal_icp .
2. Alonso, P. et al. chimie Cou..Ed. McGraw-Hill. 1990.
3. Álvarez Jiménez, MD et Gómez del Río, MI Guide didactique Chimie analytique II. UNED. 1999.
4. Arrivées Jimeno Siro; Burriel Barcelo Fernando; Hernandez Mendez Jésus; Lucena Comte Felipe. Chimie Analytique Qualitative. ISBN : 8497321405. ISB. 2006.
5. Ayres, Gilbert H. Analyse chimique quantitative. Éditions du Château, 4e éd . ISBN : 8421902806. 1981.
6. Bermejo Barrera. M del Pilar. Chimie analytique générale, quantitative et instrumentale. Paraninfo éditoriale. 7e édition. ISBN : 8428318093. 1990.
7. Blanco, M., Cerdá, V. et Sanz Medel, A., Spectroscopie atomique analytique, Publications de l' Université autonome de Barcelone. 1990.
8. Brodé. RW, Spectroscopie chimique, New York 1952.
9. Burriel, MF, Lucena, CF. Chimie Analytique Quantitative. Édition révolutionnaire. La Havane.1978.
10. Burriel, F. Chimie analytique qualitative. Paraninfo éditoriale. ISBN : 8497321405. pp 1072. , 2003.