KERNMAGNETISCHE RESONANZ (NMR)
- Details
- Germán Fernández
- KERNMAGNETISCHE RESONANZ (NMR)
- Zugriffe: 14335
Atomkerne rotieren um sich selbst (Spin) und weisen einen Drehimpuls auf, der durch den Ausdruck \begin{equation}\label{ec1} L=\sqrt{I(I+1)}\hbar \end{equation} Drehimpuls gegeben ist hängt von der Quantenzahl I (Drehimpulsquantenzahl oder Kernspin) ab, die je nach Kernart unterschiedliche Werte annehmen kann, I=0, 1/2, 1, 3/2, 2, 5/2, 3,.....
Die Berechnung der Spinquantenzahl für einen Kern erfolgt durch Addition der Spins von ungepaarten Protonen und Neutronen. Beispielsweise hat Wasserstoff I=1/2, da er nur aus einem Proton besteht.
Die erlaubten Quantenspinzustände sind durch $m_I$ gegeben, was folgende Werte annimmt \begin{equation} m_I=-I, -I+1, ...., I-1, I \end{equation} Die Anzahl der Werte, die $m_I$ für einen gegebenen Wert von I annimmt, ist $2I+1$. Für einen Kern mit I=1/2 gibt es also zwei mögliche Quantenspinzustände, gegeben durch $m_I=-1/2, 1/2$. Ein Kern mit I=1 hat drei erlaubte Spin-Quat-Zustände $m_I=-1, 0, +1$. Ohne Magnetfeld sind die Spinquantenzustände entartet.
- Details
- Germán Fernández
- KERNMAGNETISCHE RESONANZ (NMR)
- Zugriffe: 14880
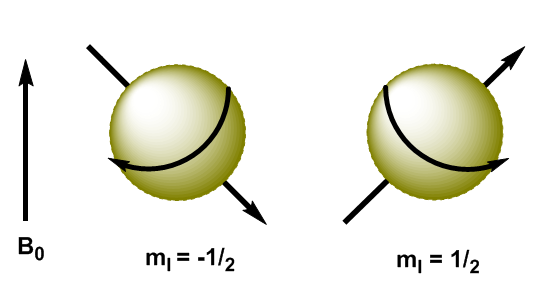
Atomkerne sind geladene Teilchen und ihr Drehimpuls erzeugt ein magnetisches Moment, dargestellt durch $\mu$ \begin{equation}\label{ec3} \mu=\gamma L = \gamma \sqrt{I(I+1)} \hbar \end{equation} wobei $\gamma$ die gyromanetische Konstante ist, die für jeden Kern charakteristisch ist. Wenn wir ein Magnetfeld, $B_0$, an Kerne mit I=1/2 anlegen, sind die magnetischen Momente so orientiert, dass die Kerne mit $m_I=1/2$ ihr magnetisches Moment mit dem Feld und dem ausgerichtet haben Kerne mit $ m_I=-1/2$ haben ihr magnetisches Moment entgegengesetzt zum angelegten Feld.
- Details
- Germán Fernández
- KERNMAGNETISCHE RESONANZ (NMR)
- Zugriffe: 11982
In der Quantenmechanik ist der magnetische Drehimpuls in z-Richtung gegeben durch $L_z=m_I\hbar$. Der Kernel mit $m_I=1/2$ hat $L_z=1/2\hbar$, während der Kernel mit $m_I=-1/2$ $L_z=-1/2\hbar$ hat. Das magnetische Moment in dieser Richtung ist $\mu_z=\gamma L_z$.
Diese Gleichungen erlauben es uns, die Energie beider Niveaus zu bestimmen. \begin{equation}\label{ec4} E=-\mu_zB_0=-\gamma m_I\hbar B_0 \end{equation} Jetzt können wir die Energiedifferenz zwischen den beiden Niveaus berechnen \begin{equation}\label{ec6} \ Delta E=-\gamma (-1/2)\hbar B_0 + \gamma 1/2\hbar B_0=\gamma\hbar B_0 \end{equation}
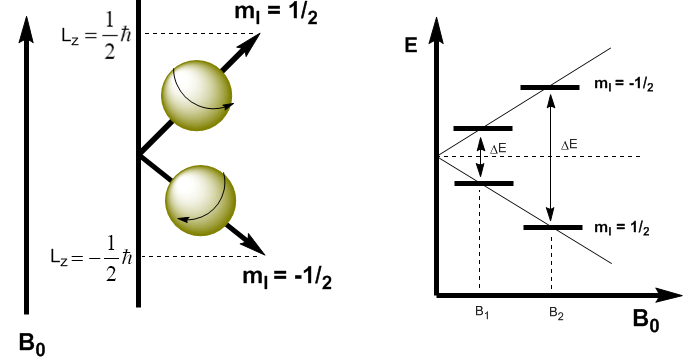
Wie in der Gleichung (\ref{ec6}) zu sehen ist, hängt die Energiedifferenz vom angelegten Magnetfeld ab. Wenn das Magnetfeld niedrig ist, ist $\Delta E$ klein und die Besetzungsdifferenz zwischen den beiden Niveaus ist ebenfalls klein, was ein Empfindlichkeitsproblem verursacht. Bei starken Magnetfeldern haben wir eine signifikante Trennung zwischen den Ebenen, was zu einem großen Populationsunterschied führt, wodurch eine größere Empfindlichkeit erreicht wird.
- Details
- Germán Fernández
- KERNMAGNETISCHE RESONANZ (NMR)
- Zugriffe: 14022
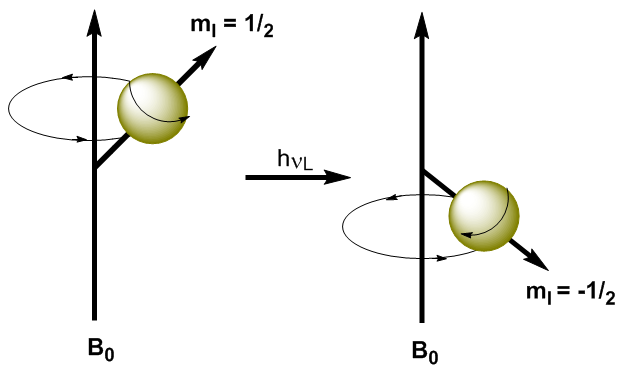
Durch Anwendung elektromagnetischer Strahlung geeigneter Frequenz (Radiowellen) ist es möglich, die Kerne vom niedrigeren Energieniveau ($m_I=1/2$) auf das höhere Energieniveau ($m_I=-1/2$) zu heben. Wenn elektromagnetische Strahlung und die Präzession des Kerns in Resonanz treten, kommt es zur Absorption. Wir können die Resonanzfrequenz (Larmor-Frequenz) mit der Planck-Gleichung berechnen. \begin{equation}\label{ec7} \Delta E = h\nu_L \end{equation} Einsetzen der Energiedifferenz in die Gleichung (\ref{ec7}) \begin{equation}\label{ec8} \gamma\ hbar B_0=h\nu_L \end{equation} Auflösen nach der Larmor-Frequenz \begin{equation}\label{ec10} \nu_L=\frac{\gamma}{2\pi}B_0 \end{equation} Wie man sieht in der Gleichung (\ref{ec10}) hängt die Frequenz, bei der der Übergang auftritt, vom angelegten Magnetfeld ab. Die Erhöhung des Magnetfelds erzeugt eine Erhöhung der Energiedifferenz zwischen den Spinniveaus, sodass eine Strahlung mit höherer Frequenz erforderlich ist, um den Übergang zu erzeugen.
- Details
- Germán Fernández
- KERNMAGNETISCHE RESONANZ (NMR)
- Zugriffe: 17430
In der Magnetresonanz werden Kerne mit einem anderen Spin-Drehimpuls als Null verwendet, wie z. B. $^1H$ und $^{13}C$. Die Resonanzfrequenzen sind jedoch nicht für alle Wasserstoff- oder Kohlenstoffkerne gleich, sie hängen von der chemischen Umgebung ab, die jeden Kern umgibt. Dies liegt daran, dass die Elektronen, die jeden Kern umgeben, ein Magnetfeld erzeugen, das dem angelegten entgegengesetzt ist, man sagt, dass die Kerne abgeschirmt sind, wobei $\sigma$ die Abschirmkonstante ist. \begin{equation}\label{ec11} B_{eff}=B_{0}-\sigma B_0=(1-\sigma)B_0 \end{equation} $B_{ef}$ ist das auf das Proton wirkende Nettomagnetfeld; $B_0$ ist das angelegte Magnetfeld; $\sigma$ ist die Rasterkonstante, unabhängig vom angelegten Feld. Unter dieser neuen Situation, bei der die Kerne durch die umgebende Elektronendichte abgeschirmt sind, wird die Resonanzfrequenz zu \begin{equation}\label{ec12} \nu=\frac{\gamma}{2\pi}(1 -\sigma)B_0 \end{equation} Kerne mit unterschiedlichen chemischen Umgebungen haben unterschiedliche Abschirmkonstanten und erzeugen unterschiedliche Signale im NMR-Spektrum.
- Details
- Germán Fernández
- KERNMAGNETISCHE RESONANZ (NMR)
- Zugriffe: 23305
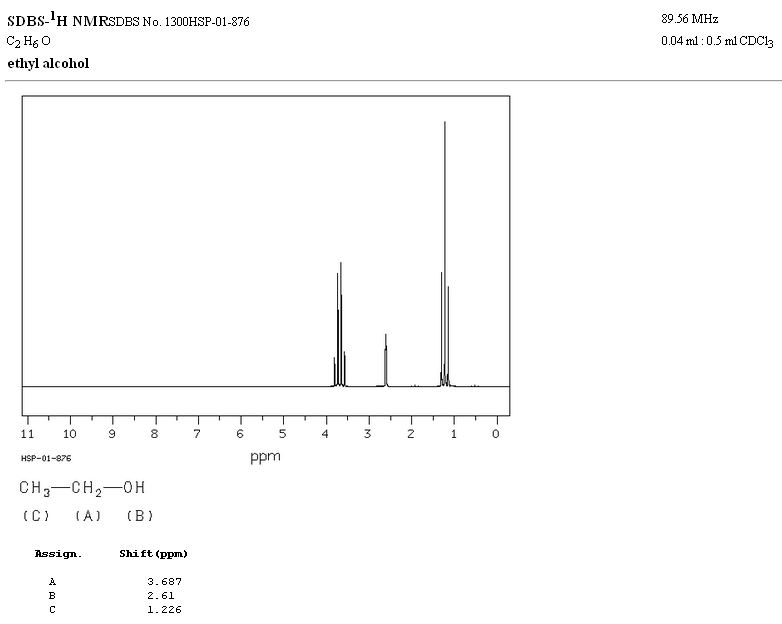
Im NMR-Spektrum von Ethanol werden aufgrund der Existenz von 3 Arten von Wasserstoffatomen mit unterschiedlichen chemischen Umgebungen drei verschiedene Signale beobachtet. Wasserstoff A ist aufgrund des Vorhandenseins von Sauerstoff (elektronegatives Atom, das Elektronendichte entfernt) unabgeschirmter als C. Die chemische Umgebung von Wasserstoff B, direkt an Sauerstoff gebunden, ist ebenfalls anders und schwingt mit einer anderen Frequenz als die vorherigen.
- Details
- Germán Fernández
- KERNMAGNETISCHE RESONANZ (NMR)
- Zugriffe: 24795
Signale im NMR-Spektrum werden auf einer vom angelegten Magnetfeld unabhängigen Skala gemessen, die als chemische Verschiebung bezeichnet und durch den Buchstaben $\delta$ dargestellt wird. Unabhängig davon, bei welchem Magnetfeld das Spektralfotometer arbeitet, erhält man die Signale einer chemischen Verbindung immer bei denselben Werten von $\delta$. \begin{equation} \delta=\frac{\nu_{sample}-\nu_{reference}}{\nu_{reference}}\cdot 10^6 \end{equation} Per Definition wird als Null der Maßstab genommen chemische Verschiebung des Signals von Tetramethylsilan ($Si(CH_3)_4$). Berechnen wir die chemische Verschiebung für $CH_3Br$ mit dem Wissen, dass in einem 90-MHz-Gerät die Absorptionsfrequenz bei 90 000 237 Hz liegt. \begin{equation} \delta =\frac{90 000 237 - 90 000 000}{ 90 000 000} \cdot 10^{6}=2,63 \end{equation} In einem Spektralphotometer, das bei 300 MHz arbeitet, tritt die Absorption bei 300 000 790 Hz auf, die Wiederholung der vorherigen Berechnung ergibt die gleiche chemische Verschiebung.\ \ Je mehr unabgeschirmte Wasserstoffe herauskommen größere Verschiebungen: $CH_3Br\rightarrow \delta =2.63$; $CH_2Br_2\rightarrow \delta =$4,90; $CHBr_3\rightarrow \delta = $6,82
- Details
- Germán Fernández
- KERNMAGNETISCHE RESONANZ (NMR)
- Zugriffe: 30207
Die folgende Tabelle zeigt die Bereiche, in denen die NMR-Signale für verschiedene Arten von Wasserstoffen erscheinen.
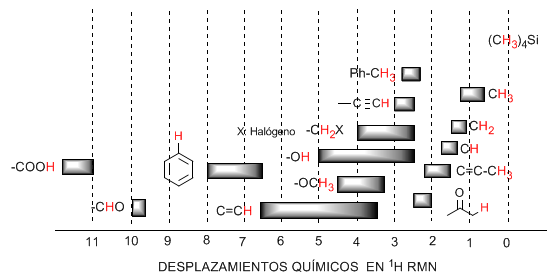
♦ Wasserstoffe, die sich an aliphatischen Ketten befinden, weisen Werte von $\delta$ nahe 1 auf. Sie steigen leicht an, wenn von primären zu sekundären oder tertiären Kohlenstoffen übergegangen wird.
♦ Allylische Wasserstoffatome liegen zwischen 1,5 und 2,1.
♦ Alpha-Wasserstoffe in Bezug auf Carbonyle und Säurederivate liegen zwischen 2 und 2,5.
♦ Die Benzylwasserstoffe zwischen 2,3 und 2,7.
♦ Der Wasserstoff von terminalen Alkinen zwischen 2,5 und 3.
♦ An Kohlenstoffe gebundene Wasserstoffe mit Halogenen zwischen 2,5 und 4, abhängig von der Elektronegativität des Halogens
♦ Die Wasserstoffatome der Hydroxylgruppe zwischen 2,5 und 5. Sehr großer Bereich aufgrund der Bildung von Wasserstoffbrückenbindungen.
♦ Wasserstoffe mit sauerstoffgebundenen Kohlenstoffen vom Ethertyp zwischen 3,3 und 4,5.
♦ Olefine Wasserstoffatome zwischen 3,5 und 6,5.
♦ An aromatische Systeme gebundene Wasserstoffe zwischen 6,5 und 8.
♦ Wasserstoff aus Aldehyden 9.5-10
♦ Wasserstoff aus der Carbonsäuregruppe über 11.
- Details
- Germán Fernández
- KERNMAGNETISCHE RESONANZ (NMR)
- Zugriffe: 15787
Elektronegative Substituenten entfernen die Elektronendichte, heben die Abschirmung der Wasserstoffatome auf und verschieben das Signal zu hohen δ$-Werten.
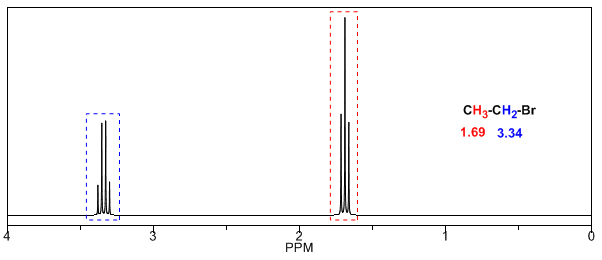
In der folgenden Tabelle sehen Sie den Einfluss verschiedener Atome auf das Signal der Methylwasserstoffe.
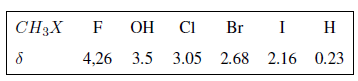
Wie aus der Tabelle ersichtlich ist, ist die chemische Verschiebung umso größer, je größer die Elektronegativität der Gruppe X ist.
Die Wirkung der elektronegativen Gruppen ist additiv, je größer die Anzahl der Gruppen ist, desto ungeschirmter ist das Proton und desto größer ist die Verschiebung
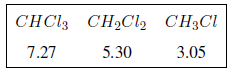
Der induktive Effekt breitet sich entlang der Ketten aus und nimmt mit zunehmendem Abstand ab, wie aus der folgenden Tabelle ersichtlich ist.
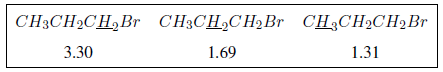
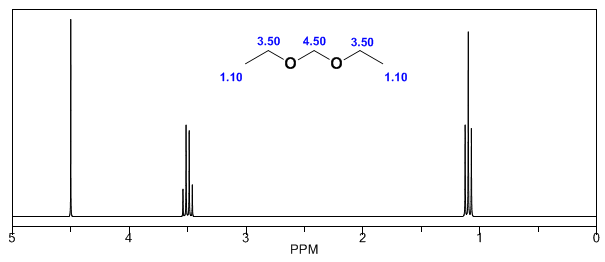
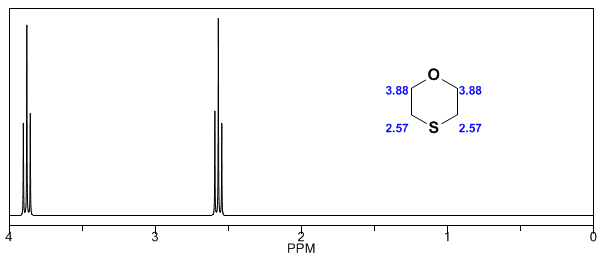
In den folgenden Spektren können die oben erwähnten Effekte auf chemische Verschiebungen beobachtet werden.
- Details
- Germán Fernández
- KERNMAGNETISCHE RESONANZ (NMR)
- Zugriffe: 23728
Die Protonen in der Nähe von Doppelbindungen und aromatischen Ringen sind aufgrund des durch die elektronischen Ströme dieser Systeme induzierten Magnetfelds besonders ungeschirmt. Das induzierte Feld wird zum angelegten addiert, wodurch eine größere Verschiebung als erwartet entsteht.
Im folgenden Bild sehen wir die elektronische Zirkulation (fettgedruckte Kurven) und das induzierte Magnetfeld (gestrichelte Linien) für ein Alken und ein Carbonyl. Beobachten Sie, wie im Bereich des Protons das induzierte Magnetfeld die gleiche Richtung und Richtung hat wie das angelegte.
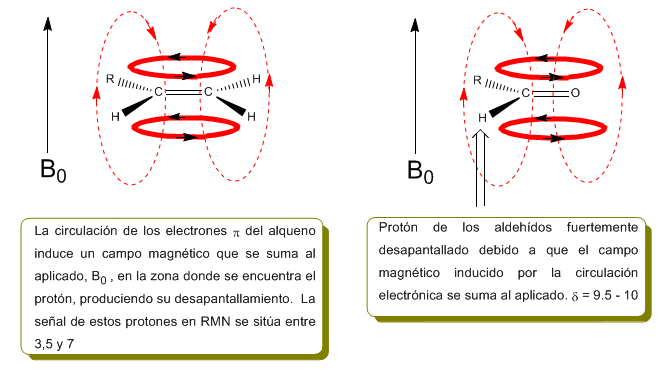
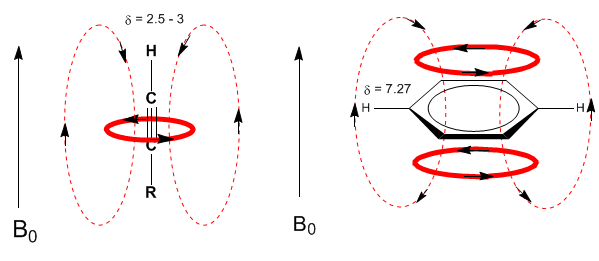
Eine ähnliche Situation wird im Fall von Benzol beobachtet. In Alkinen induziert die elektronische Zirkulation jedoch ein Magnetfeld, das dem im Protonenbereich angelegten entgegengesetzt ist. Die acetylenischen Wasserstoffatome werden mit Signalen im NMR-Spektrum bei geringen Offsets abgeschirmt.
Als nächstes füge ich einige Spektren von Alkenen, Alkinen und Aromaten hinzu.
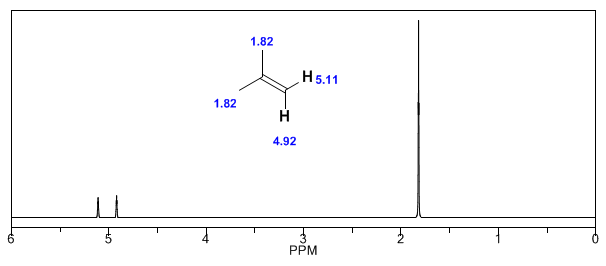
Beobachten Sie, wie die induzierten Felder die Verschiebungen des olefinischen Protons erheblich erhöhen, wobei auch die allylischen Positionen beeinflusst werden.
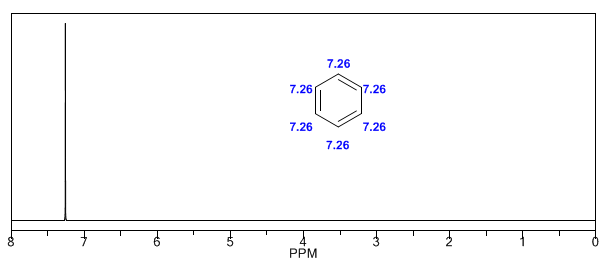
Die aromatischen Wasserstoffatome sind aufgrund des durch die Ringströme induzierten Feldes stark ungeschirmt.
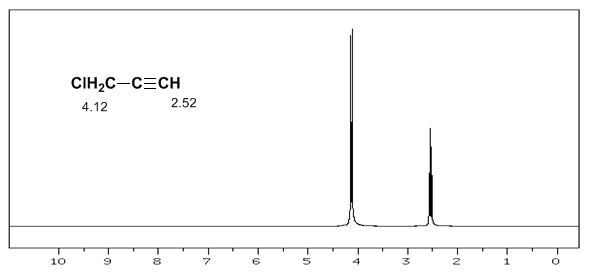
Der acetylenische Wasserstoff hat eine geringe Verschiebung, weil die Ströme ein Magnetfeld erzeugen, das dem angelegten entgegengesetzt ist.
- Details
- Germán Fernández
- KERNMAGNETISCHE RESONANZ (NMR)
- Zugriffe: 13035
Die Bildung von Wasserstoffbrückenbindungen führt zu höheren Verschiebungswerten. Das Vorhandensein von saurem Wasserstoff kann durch Zugabe von deuteriertem Wasser nachgewiesen werden, was den Austausch von saurem Wasserstoff gegen Deuterium bewirkt, wodurch das Signal verschwindet. \begin{equation} CH_3OH + D_2O \rightleftharpoons CH_3OD + HDO \end{equation} Die häufigsten sauren Wasserstoffverschiebungen in organischen Molekülen sind:
- Carbonsäuren (RCOOH) $\delta$ = 10 - 12 ppm
- Amine ($R-NH_2$) $\delta$ = 0,5 - 5 ppm
- Amide ($RCONH_2$) $\delta$ = 5-8 ppm
- Alkohole (ROH) $\delta$ = 0,5 - 5 ppm
- Phenole (Ph-OH) $\delta$ = 4 - 7 ppm
- Details
- Germán Fernández
- KERNMAGNETISCHE RESONANZ (NMR)
- Zugriffe: 27580
Die Strukturinformationen der NMR stammen aus zwei Faktoren: den unterschiedlichen beobachteten Verschiebungen in Abhängigkeit von der chemischen Umgebung, die das Proton umgibt, und der Kopplung zwischen den Spins benachbarter Protonen, die die Aufspaltung der Signale bewirkt.
Obwohl einige Signale im Spektrum einzelne Peaks sind, ist es üblich, Signale zu finden, die aus mehreren sehr nahen Peaks bestehen, die mit der folgenden Notation benannt werden: Singulett (s), Dublett (d), Triplett (t), Quadruplett (c) , Quintole (q), Sextole (sx) und Septole (sp) werden komplexe Signale als Multipletts bezeichnet. Der Wert von $\delta$ dieser Signale wird ihrem Zentrum zugeordnet, es sei denn, das Multiplett ist unregelmäßig, in diesem Fall wird das Intervall angegeben.
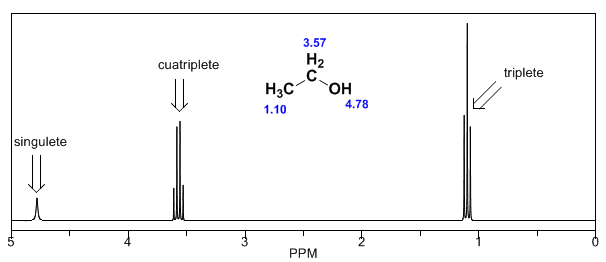
Im Spektrum von Ethanol ist ersichtlich, dass der Hydroxyl-Wasserstoff ein Singulett erzeugt, das Wasserstoffpaar an Kohlenstoff eins ein Quadruplett erzeugt und die drei Wasserstoffatome an Kohlenstoff zwei ein Triplett erzeugen.
Erklärung der Spin-Spin-Kopplung.
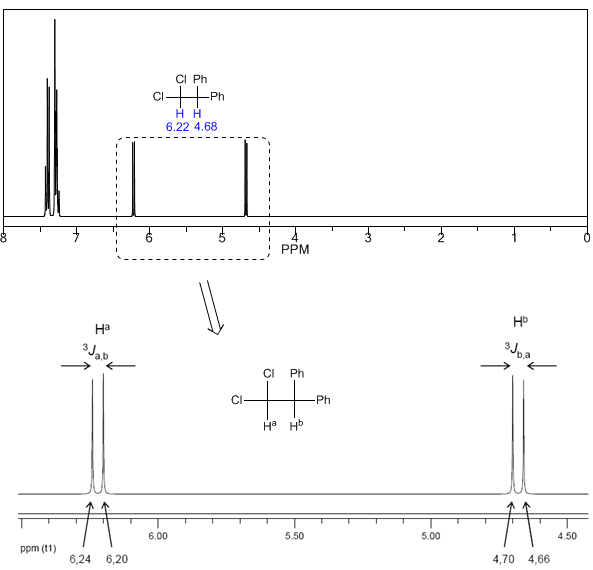
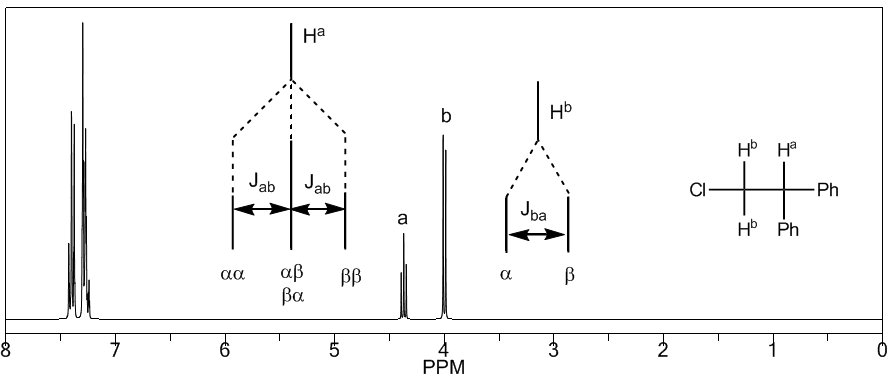
Um die Signalaufspaltung durch Spin-Spin-Kopplung zu verstehen, untersuchen wir das Spektrum von 1,1-Dichlor-2,2-diphenylethan ($Cl_2CH^{a}CH^{b}Ph_2$).
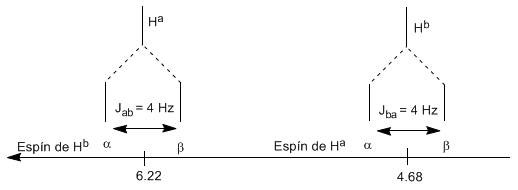
Das einem Magnetfeld $B_0$ ausgesetzte Proton $H^a$ erzeugt ein Signal bei $\delta_a=6,22 ppm$. Das Proton $H_b$ erzeugt jedoch ein kleines Magnetfeld, das auf das Proton $H_a$ einwirkt. Bei etwa der Hälfte der Moleküle ist das $H_b$-Proton auf das angelegte Feld ausgerichtet (Alpha-Spin) und bei der anderen Hälfte gegen das Feld ausgerichtet (Beta-Spin). Wenn $H_b$ den Spin $\alpha$ hat, wird $H_a$ einem etwas größeren Feld ausgesetzt und schwingt mit einer höheren Frequenz ($\delta$ etwas höher). Wenn $H_b$ den Spin $\beta$ hat, wird $H_a$ einem etwas kleineren Feld ausgesetzt und schwingt bei einer niedrigeren Frequenz ($\delta$ etwas niedriger), was die Aufspaltung der anfänglichen Spitze in zwei durch a getrennte Signale erzeugt Abstand von 4 Hz, genannt Kopplungskonstante (J). Dieselbe Überlegung kann man für das Proton $H_b$ anstellen.
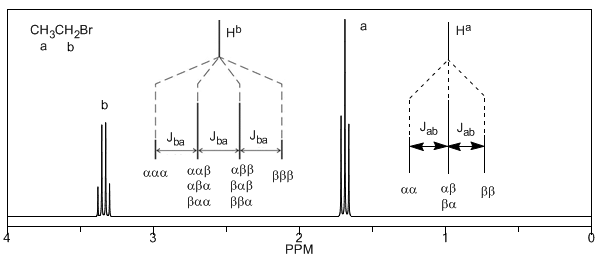
Als nächstes untersuchen wir die Kopplung eines Wasserstoffatoms $H^a$, das zwei benachbarte Wasserstoffatome $H^b$ hat. In dieser Situation wird die Aufspaltung des Wasserstoffsignals $H^a$ in drei Peaks (Triplett) beobachtet, wobei der zentrale Peak doppelt so intensiv ist wie die äußersten. Die Wasserstoffatome $H^b$ koppeln ihrerseits mit $H^a$ und erzeugen zwei Peaks gleicher Intensität (Dublett).
Schließlich diskutieren wir die Kopplung eines Protons mit drei äquivalenten Nachbarprotonen. In diesem Fall wird ein Signal beobachtet, das aus vier Peaks (Quadtriplet) besteht. Die zentralen Spitzen sind dreimal so intensiv wie die äußersten Spitzen.
- Details
- Germán Fernández
- KERNMAGNETISCHE RESONANZ (NMR)
- Zugriffe: 17746
Aus der obigen Diskussion kann abgeleitet werden, dass ein Proton ein Signal mit einer Anzahl von Peaks erzeugt, die um eine Einheit größer ist als die Anzahl benachbarter Wasserstoffatome. Im folgenden Bild sehen wir die Peaks, die von einem Wasserstoff $H^b$ erzeugt werden, wenn er mit einer Reihe von variablen Wasserstoffen $H^a$ gekoppelt wird
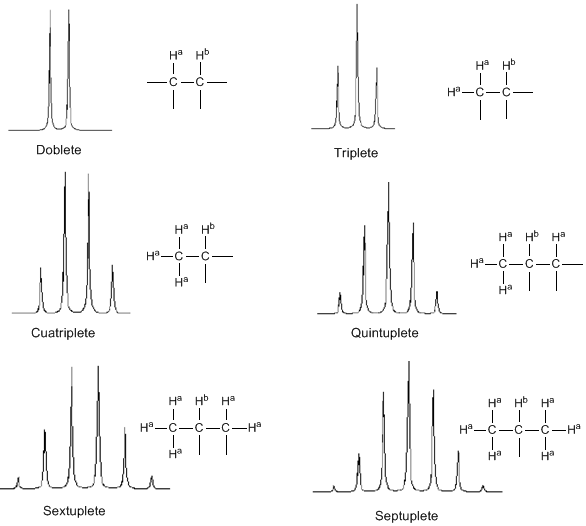
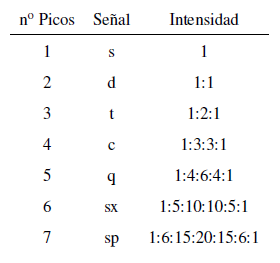
Die Intensität der Spitzen eines Signals wird durch das Pascalsche Dreieck (Tartaglia) angegeben.
Bei der Anwendung der N+1-Regel sind zwei Überlegungen zu berücksichtigen:
- In Molekülen des Typs $A-CH2^{a}-CH2}^{b}-CH2}^{a}-A$ treten die $H^b$-Protonen als Quintuple auf.
- In Molekülen des Typs $A-CH_2-CH_2-A$ sind die vier Protonen äquivalent und ergeben ein Singulett.
- Details
- Germán Fernández
- KERNMAGNETISCHE RESONANZ (NMR)
- Zugriffe: 12060
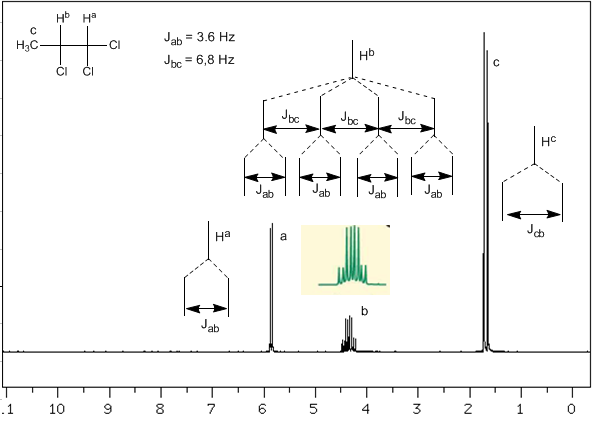
Wir werden nun eine komplexere Situation analysieren, in der die Kerne $H^b$ unterschiedliche Kopplungskonstanten mit den benachbarten Kernen $H^c$ und $H^a$ aufweisen. Da wir wissen, dass die Kopplungskonstanten zwischen diesen drei Kernen $J_{ab}=3.6\;Hz$ und $J_{bc}=6.8\;Hz$ sind, koppeln wir es zuerst mit $, um das Signal von $H^b$ zu erhalten H^c$ (größte Kopplungskonstante), was vier Peaks ergibt (N+1-Regel), die dann mit $H^a$ gekoppelt werden, wobei sich jeder in zwei Peaks aufspaltet. Insgesamt wird ein aus acht Spitzen bestehendes Signal erhalten.
- Details
- Germán Fernández
- KERNMAGNETISCHE RESONANZ (NMR)
- Zugriffe: 14645
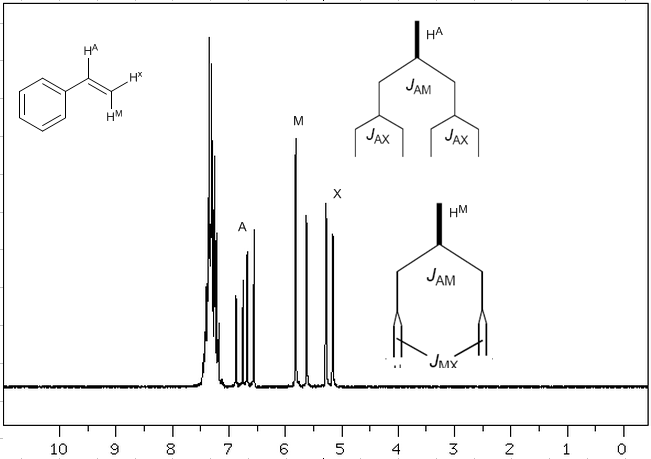
Berücksichtigt man, dass die Reihenfolge der Kopplungskonstanten in Alkenen $J_{trans}>J_{cis}>J_{geminal}$ ist, sind die Kopplungsbäume für die Wasserstoffatome $H^a$ und $H^M$ wie auf angegeben Das Spektrum. Könntest du den Baum für $H^x$ zeichnen?