Stereochemistry and conformational analysis
Stereochemistry is the branch of chemistry that deals with the three-dimensional aspects of molecules and their reactivity. Organic Chemistry cannot be understood without considering Stereochemistry. Biological systems are highly selective and often discriminate molecules with very small stereochemical differences.
Conformational analysis is part of the spatial study of molecules, that is, of Stereochemistry.
CONSTITUTION, CONFIGURATION AND CONFORMATION
Compounds with the same formula are called isomers.
empirical but different structure.
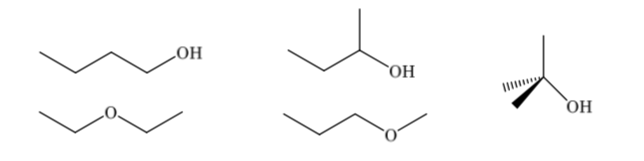
The constitutional isomers differ in their binding sequence, they present different connectivity. A compound with the molecular formula C4H10O can have different constitutions:
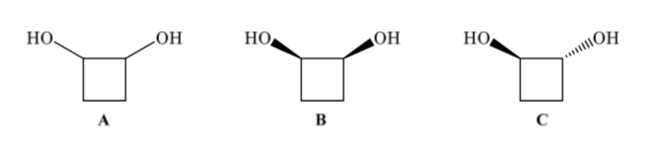
Configurational isomers have the same connectivity but differ in the spatial arrangement of the atoms. 1,2-Cyclobutanediol (A) can have two configurational isomers depending on whether the two OH groups are on the same side (B) or on opposite sides (C)
Conformational Isomers have the same constitution, the same configuration, but they differ spatially in that they pass from one isomer to another by simple rotation of a bond. The various shapes that molecules acquire as a result of rotation around a single bond are called conformations, each of which is a conformer.
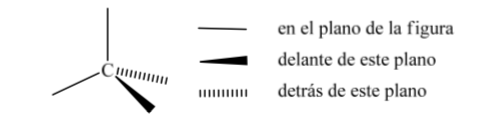
One of the most widely used representations of carbon tetrahedron is the so-called perspective representation.
Thus, in the case of chloroethane we can represent the different conformers in perspective as follows:
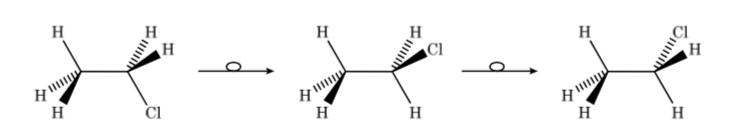
Configurational and conformational isomers are stereoisomers. Stereoisomers have the same connectivity but differ in the orientation of some of their atoms in space. They often present considerable differences in their physical, chemical and biological properties.
CONFORMATION IN ACYCLIC ORGANIC MOLECULES
Representation of organic molecules
There are several ways to represent molecules in the plane.
Representation in Perspective or Easel
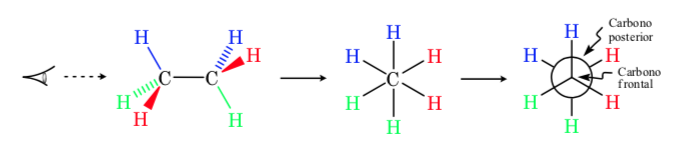
Newman projection
In a Newman projection, two adjacent carbon atoms in a molecule are depicted inside a circle. At the front is the carbon atom seen from the front and behind the circle is the overlapping carbon atom. The three substituents of each carbon come out from the center of the circle in front and behind.
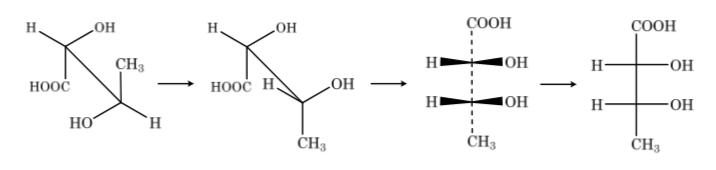
Fisher representation
In the Fischer representation, a tetrahedral carbon atom is represented by the center of a cross and the substituents are placed at the ends in such a way that the vertical lines indicate that the bonds are directed towards the back of the plane of the paper and the horizontal ones towards the front. . The main chain is arranged vertically.
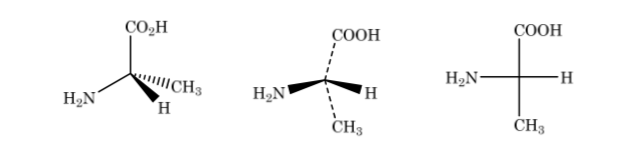
As shown above the representation of 2-aminopropanoic acid
If the compound has different carbon atoms, the carbon chain is drawn vertically with the substituents of each carbon horizontally, always maintaining the rule that the vertical bonds go behind the plane and the horizontal ones forward.
Ethane conformations
The σ bond that joins the carbon atoms in ethane is cylindrically symmetric and allows free rotation around it. Rotation around the C–C bond interconverts the different conformations of ethane. The study of its thermodynamic and kinetic behavior is called Conformational Analysis.
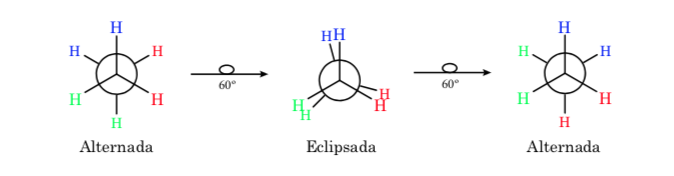
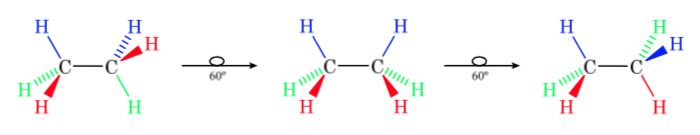
The first figure represents the ethane molecule where the hydrogen atoms are as far apart as possible with minimal interaction between them. It is called the alternate conformation.
The second comes from having made a 60o turn to the C–C bond and where the hydrogen atoms are eclipsed.
Again, a 60o turn to the C–C bond leads to an alternate conformation.
The potential energy of the molecule is minimal for the staggered conformation, increases with rotation, and reaches a maximum for the eclipsed conformation. The energy difference or energy barrier to overcome to pass from one conformation to another is of the order of 3 kcal (see drawing).
Most ethane molecules exist naturally in the most stable conformation. Since the energy barrier is not very high at room temperature, the number of collisions with sufficient energy is large enough that conformer interconversion is fast. The energy required to rotate the molecule about the C–C bond is due to torsional strain.
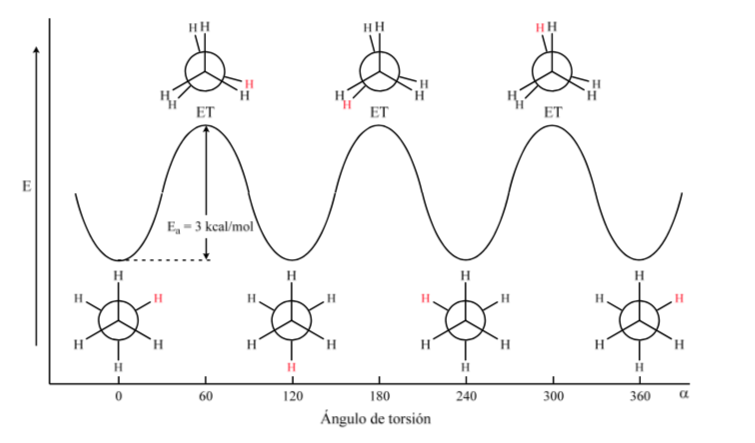
The above diagram represents the energy changes when going from one conformation to another during the rotation of the C–C single bond.
When the ethane hydrogens are replaced by other atoms or groups, the energy levels of the conformations will change and their study will have to be carried out for each case.
propane conformations
In the case of the propane molecule, if we represent the Newman projections according to one of the C – C bonds and we rotate the bond, we find ourselves, as in the case of ethane, with eclipsed and alternate conformations.
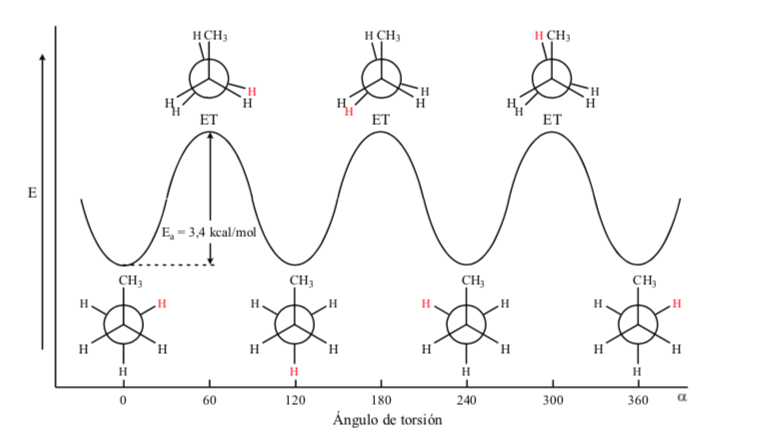
The energy barrier between an alternate and an eclipsed form is greater than in the case of ethane (3.4 kcal mol-1) since the methyl group is larger than hydrogen and the interactions are greater. Staggered conformations are more stable than eclipsed conformations.
Conformations of n-Butane
If we look at the central C–C bond, we can consider butane when representing it in Newman as a molecule similar to ethane in which two hydrogen atoms have been replaced by two methyl groups.
As in ethane, the alternate conformations have lower energy and are more stable than the eclipsed ones: 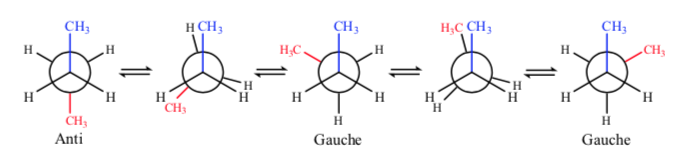
Of all the alternate conformations, the one with the two methyl groups in the most opposite position possible (dihedral angle 180o) is called anti conformation and is the most stable. The other two alternate conformations are of similar energy to each other and are called gauche conformations where the methyl groups meet at a dihedral angle of 60o to each other. Two such conformations exist depending on the rotation around the C–C bond.
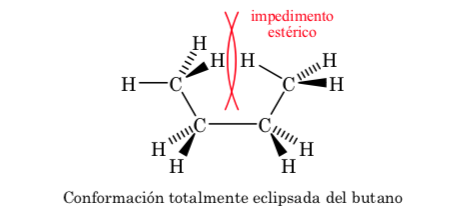
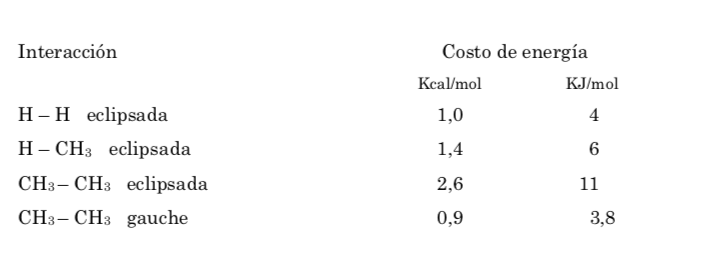
Of the three eclipsed conformations, we first highlight the one where the two methyl groups are eclipsed, which generates greater interaction and therefore less stability. The other two eclipsed conformations have lower energy content and are somewhat more stable. See the energy diagram.
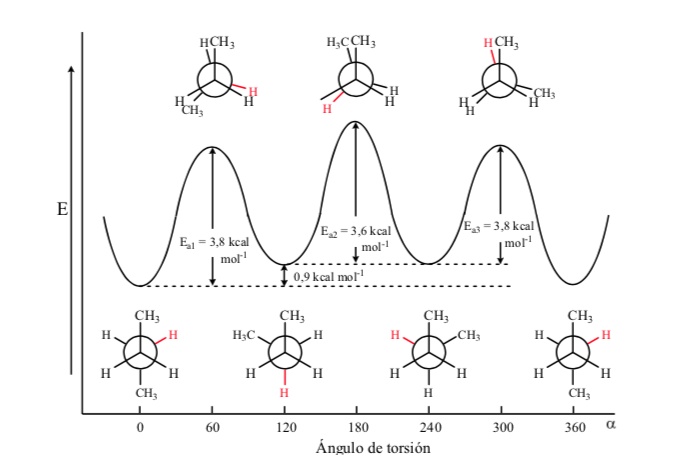
Collisions provide the energy needed to overcome the activation energy barrier.
CONFORMATION IN CYCLIC ORGANIC MOLECULES
Cycloalkanes are subjected to a ring strain called Bayer strain due to their cyclic structure. This tension is the result of three factors:
- Link Voltage
- Eclipse of atoms and bonds - Steric Strain
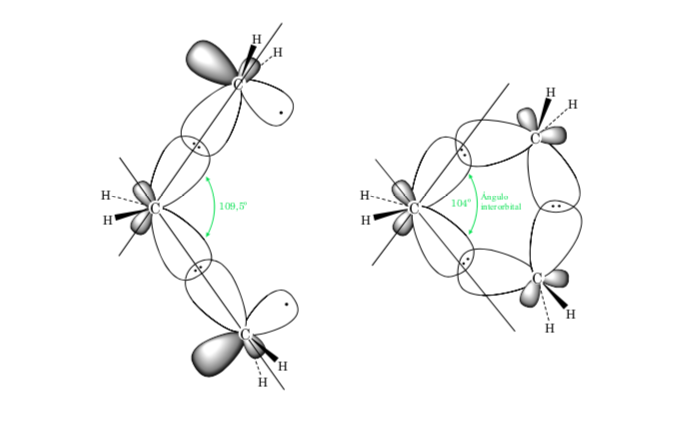
A. Bond Strain in cycloalkanes is due to the difference in orbital overlap with respect to an acyclic alkane. The closer the value of angle C–C–C is to the tetrahedral (109.5o), the greater the overlap and the lower the bond tension.
The steric effect is due to the interaction of atoms through space. To avoid that the interaction between the atoms is high, the molecule acquires a preferential conformation, this is the most stable. To reduce stresses and thus achieve stability, the preferred conformation will be one in which the bulky groups are as far apart as possible.
CONFORMATION IN CYCLES OF THREE, FOUR AND FIVE CARBON ATOMS
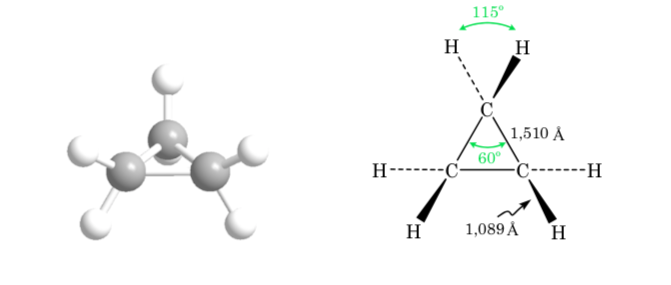
cyclopropane
The deviation of the bond angle of the three-membered ring from the angle corresponding to sp3 hybridization is large, so cyclopropane has a very high angle strain.
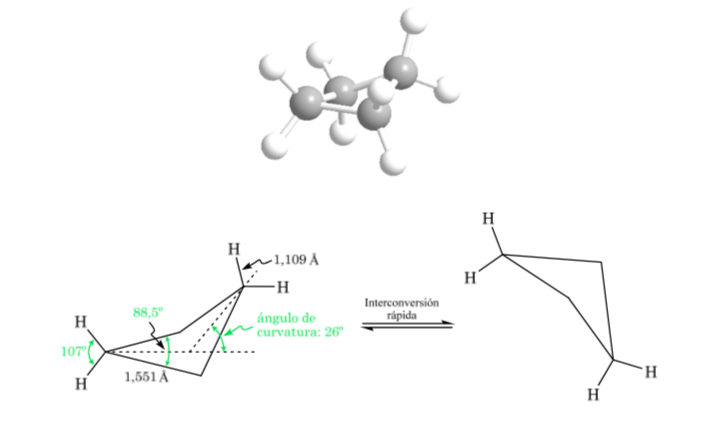
Cyclobutane: Butterfly conformation
The molecule has a slightly warped structure. The C–C bonds are less curved and their reactivity is less than in the case of cyclopropane. The interconversion between the different conformations is rapid.
Cyclopentane: Conformation on
Cyclopentane, if it were planar, would have a high eclipsement strain, due to ten eclipsed H–H interactions, resulting in a folding in the ring. The folding releases torsional stress. Cyclopentane does not exhibit the reactivity of the three- and four-membered rings.
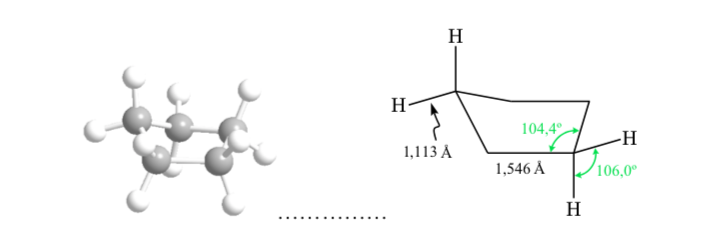
CONFORMATIONAL ANALYSIS OF CYCLOHEXANES : Chair conformation
If cyclohexane had a planar structure, it would have a bond angle of 120o. A strain-free, sp3-hybridized carbon has a tetrahedral angle of 109.5o, which causes cyclohexane to assume a non-planar and more stable conformation: Chair conformation.
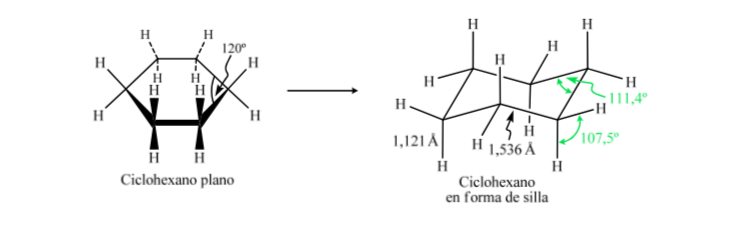
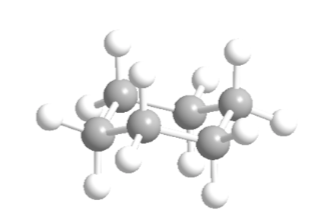
Cyclohexane in chair conformation has 6 axial hydrogen atoms and 6 equatorial hydrogens. The axial hydrogens are directed up and down the ring, while the equatorial hydrogens are directed out of the ring.
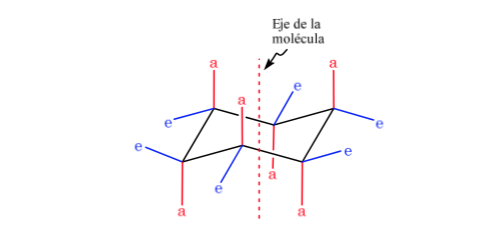
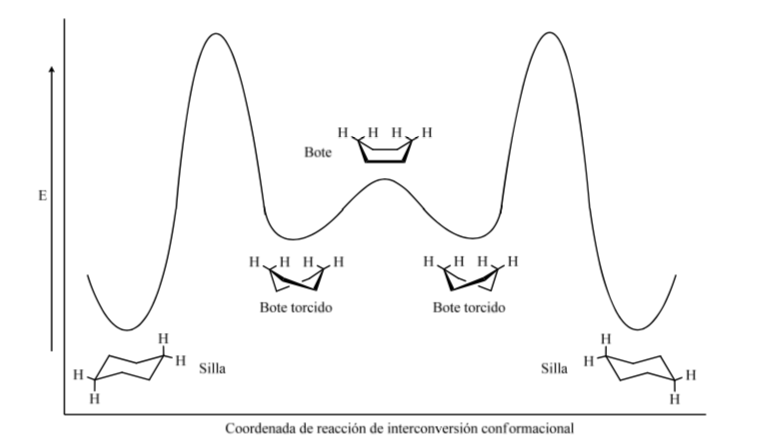
Cyclohexane in chair conformation is susceptible to modify its conformation by another chair of equal stability. This phenomenon is called interconversion of two chair forms.
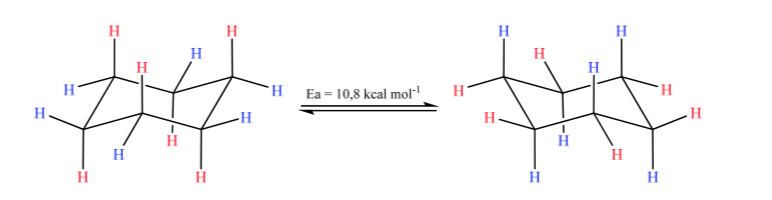
Looking closely at the interconversion, it is observed that the axial C–H bond becomes equatorial and vice versa, the equatorial becomes axial.
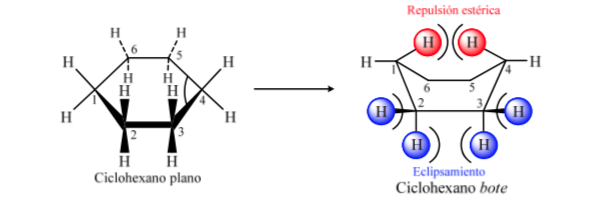
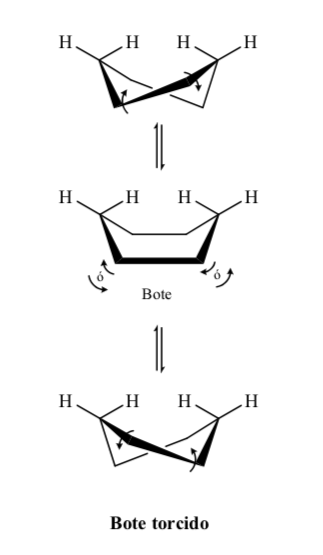
Energetic levels of the cyclohexane conformers:
If we observe a chair conformation of cyclohexane such that the C-2 atom overlaps the C-3 atom and the C-6 atom overlaps the C-5 we can draw a Newman projection of cyclohexane:
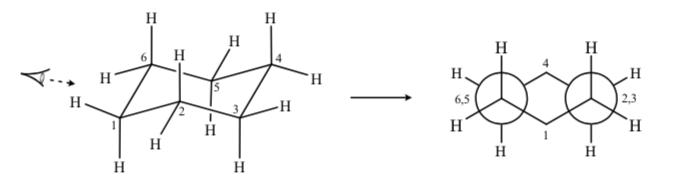
SUBSTITUTED CYCLOHEXANES
If we consider methylcyclohexane, we can propose two structures that represent it depending on whether the methyl group is in an axial or equatorial position.
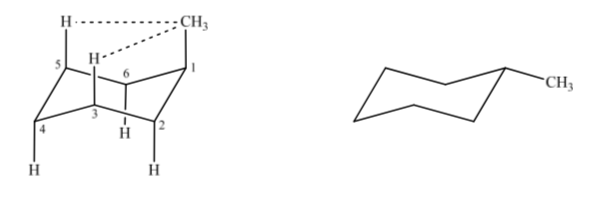
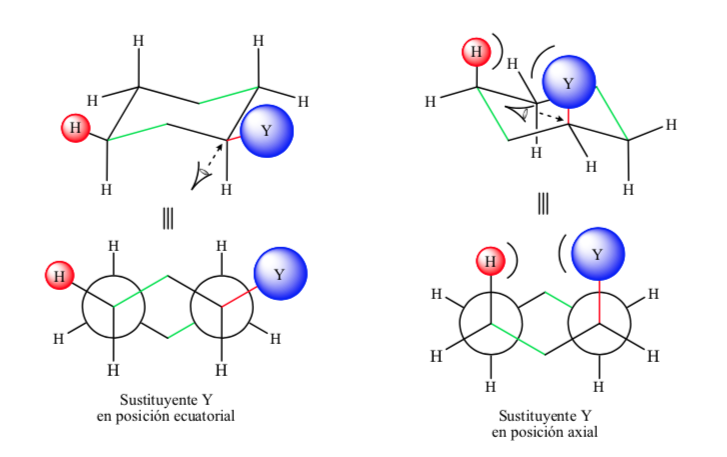
If the non-hydrogen substituent is in an axial position, interactions with the hydrogens in position 3 and 5 of the ring occur due to spatial proximity. If it is in an equatorial position, the substituent is directed out of the ring and the interactions are smaller. As expected, the conformation with the equatorial substituent is found to be about 1.8 kcal more stable than if the substituent is in the axial position. Thus it is said that a substituent in an axial position presents a 1,3-diaxial interaction that makes it less stable than in an equatorial position.
The following figure represents the Newman the two conformations of a substituted cyclohexane with both the substituent in the equatorial and axial positions.
Disubstituted cyclohexanes
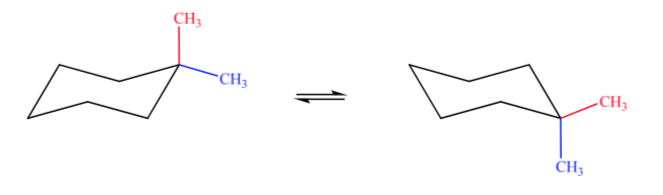
A. 1,1-dimethylcyclohexane B. 1,2-dimethylcyclohexane C. 1,3-dimethylcyclohexane D. 1,4-dimethylcyclohexane
In case A, one methyl group will be in the axial position and the other in the equatorial position, so the two possible conformations will be equally stable.
In each of the remaining cases we can represent two different structures depending on whether the two methyls are in cis (directed towards the same side of the plane) or in trans (directed towards opposite sides).
In B, the cis isomer must have both methyls on the same side of the central plane, so one methyl will be axial and the other equatorial.
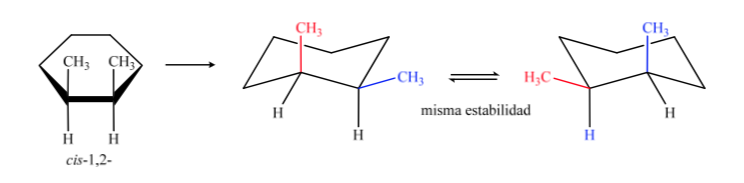
The trans isomer is the one with the two methyls facing opposite sides, so it will present both methyls in an axial position or both in an equatorial position. The conformation that both present in equatorial is more stable than in axial, therefore we can say that the trans-1,2-dimethylcyclohexane isomer is more stable than the cis-1,2-dimethylcyclohexane isomer.
In the case of 1,3-dimethylcyclohexane we can represent:
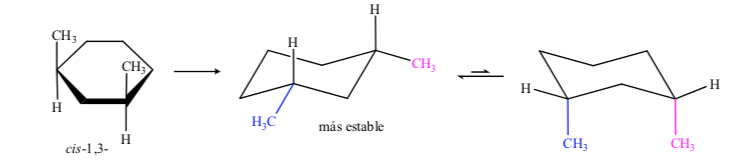
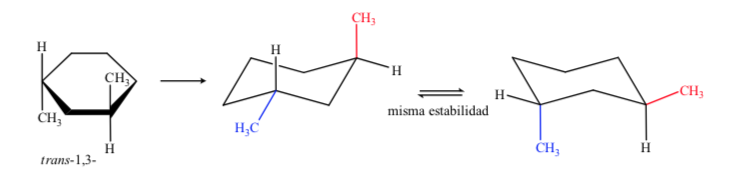
It is concluded that the cis isomer, which presents the two substituents in equatorial, is more stable than the trans, which will always have one methyl in axial and another in equatorial.
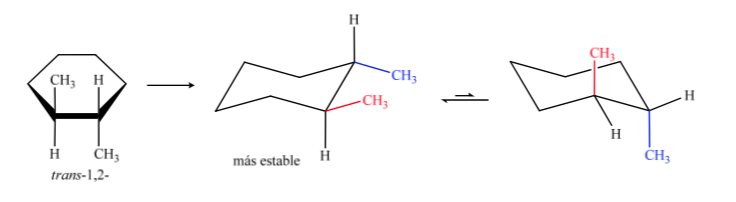
In case D, 1,4-dimethylcyclohexane we will have:
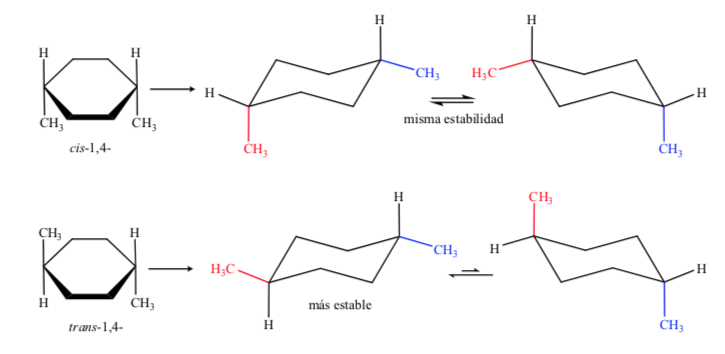
In this case, the trans isomer is more stable because the two methyls are arranged in equatorial.
When dealing with disubstituted cyclohexanes with different groups, the most stable isomer will be the one with the largest equatorial group.
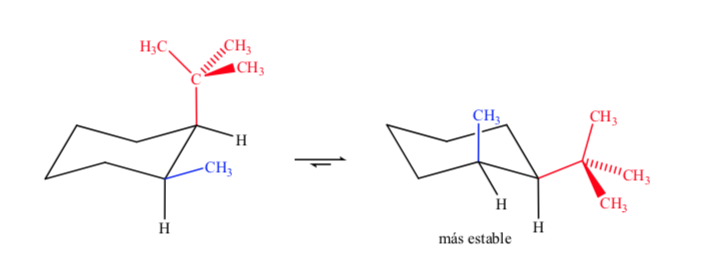
Finally, in the case of polysubstituted cyclohexanes that have different substituents, the preferred conformation will be the one with the greatest number of bulky groups in the equatorial position.
CONFORMATION IN SIX-MEMBED HETEROCYCLES:
ANOMERIC EFFECT
In organic chemistry, the anomeric effect or Edward-Lemieux effect is a stereoelectronic effect that describes the tendency of heteroatomic substituents adjacent to a heteroatom in a cyclohexane ring to prefer the axial orientation rather than the less covered equatorial orientation, which would be expected. from steric considerations. This effect was originally observed in pyranose rings by JT Edward in 1955; at that time, N.-J. Chii and Raymond U. Lemieux began to study the anomerization equilibrium of fully acetylated derivatives of some aldohexopyranoses. The term "anomeric effect" was introduced in 1958.
Some of the most common five- and six-membered heterocycles in biological processes are:
A. Oxygenated heterocycles:
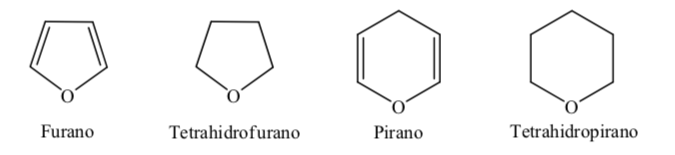
B. Nitrogenous heterocycles:
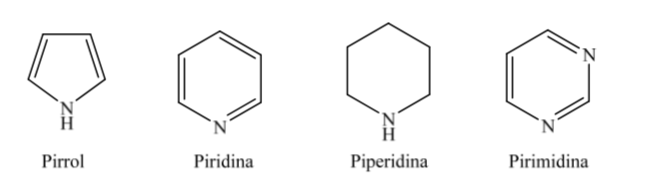
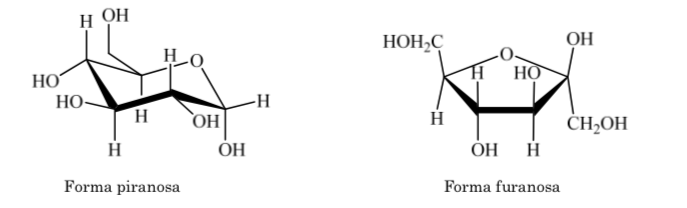
When it comes to fully hydrogenated heterocycles with five and six members such as tetrahydrofuran and tetrahydropyran, the most stable conformation they present is similar to that of cyclopentane and cyclohexane respectively, that is, conformation on or conformation chair.
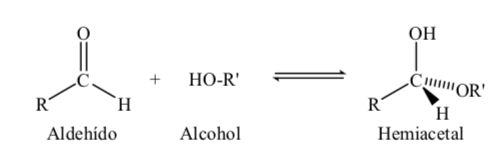
Sugars with 5 and 6 carbon atoms acquire a cyclic structure when a hemiacetal bond is formed by reaction of the carbonyl (sugar aldehyde or ketone) with a hydroxyl group.
The formation reaction of the hemiacetal can be schematized as follows:
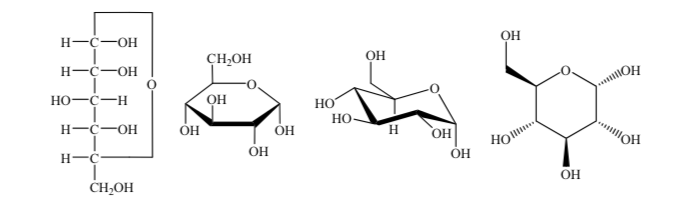
For their study, sugars are represented by linear structures following the rules of Fischer projections.
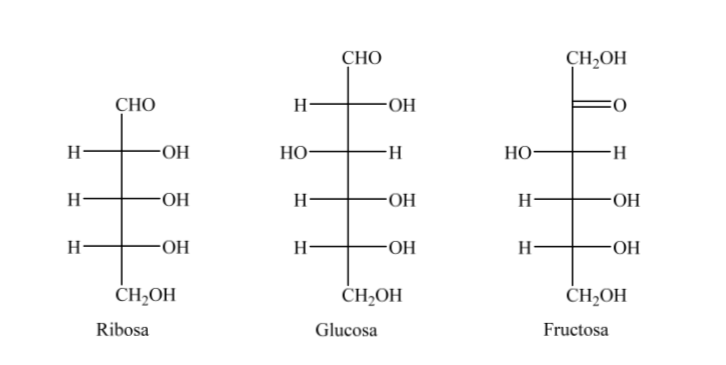
Thus, as an example, we can represent ribose, glucose and fructose:
When the hemiacetal is formed, the molecule acquires a curved arrangement in which the aldehyde function of the aldose on carbon 1 is located close to the hydroxyl on carbon 5, adding to the carbonyl double bond, generating the six-membered cyclic hemiacetal (pyranoside ring ).
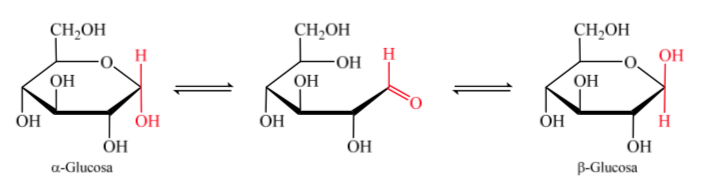
The same occurs with ketoses, but in this case the hemiacetal bond occurs between carbon 2 and carbon 5, forming a five-membered ring. (furanoside ring).
Different ways of representing the structure of glucose in cyclic form are used:
anomers
The isomers of monosaccharides with more than 5 carbon atoms that have developed a hemiacetal bond are called anomers, which has allowed them to take on a cyclic structure and determine 2 different positions for the hydroxyl group (α or β) in the new center created. .
The pyranose ring should be similar to that of cyclohexane and exist in the chair conformation in preference to the twist boat conformation to minimize torsional stresses. X-ray analysis shows these assumptions to be correct.
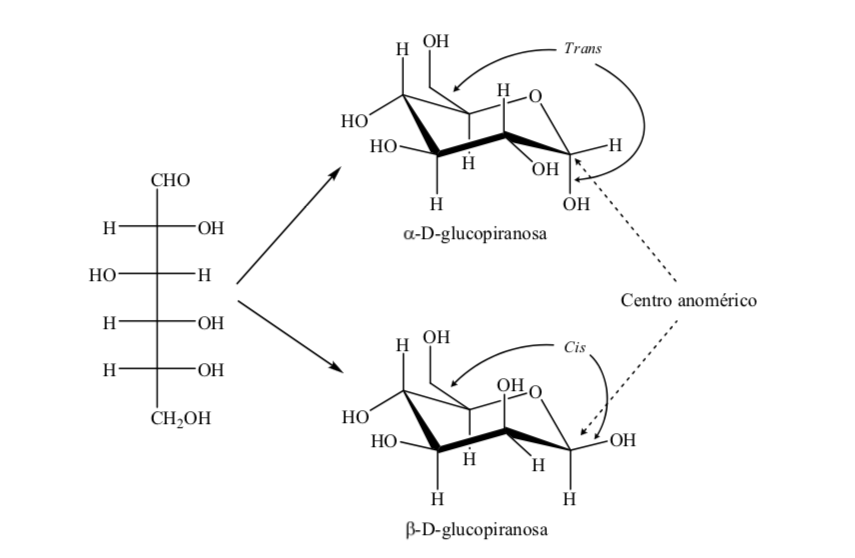
The two anomers are designated alpha (α) or beta (β) anomers, according to the configurational relationship between the anomeric center and the anomeric reference atom. The anomeric center is on the hemiacetal carbon and is the anomeric carbon C-1, which is bonded through oxygen to C-5, which is bonded to hemiketal oxygen.
There are, however, two possible chair conformations for the same D-(+)-glucopyranose anomer.
Thus, for β-D-(+)-glucopyranose there are two possible conformations. The one that presents the most voluminous groups in equatorial (A) will be more stable.
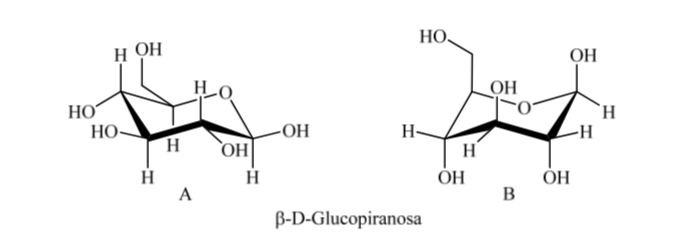
What happens for α-D-(+)-glucose? In this case, the most stable conformation is the one in which the OH on the anomeric carbon is axial and the bulky groups are equatorial (A).
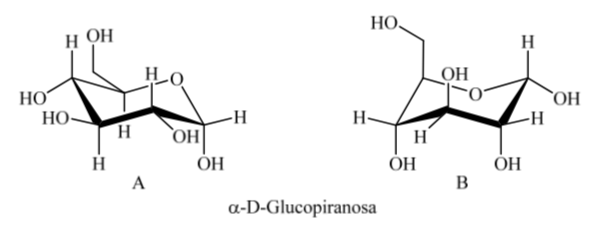
It is interesting to note that of all the D-aldohexoses, only β-D-(+)-glucopyranose can assume a conformation in which each of its large groups can occupy an equatorial position. This is consistent with the fact that β -D-(+)-glucopyranose is the sugar with the highest occurrence in nature.
electronic effects
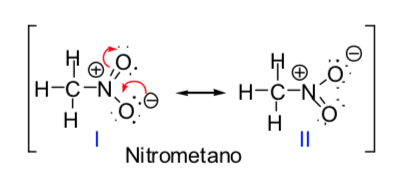
Some organic molecules can be represented by two or more Lewis structures, which differ from each other only in the placement of the electrons and are called s-resonant structures. In these cases, the molecule will have characteristics of both structures, and the molecule is actually said to be a resonance hybrid of the resonant structures. The resonance method allows us to know, in a qualitative way, the stabilization that a molecule can achieve by electronic delocalization. The greater the number of resonant structures by which a chemical species can be described, the greater its stability.
The concept of resonant structures can be applied in the description of nitromethane, which can be represented by the two Lewis structures given below:
Conjugate bonds and their energy
Conformational analysis of aromaticity
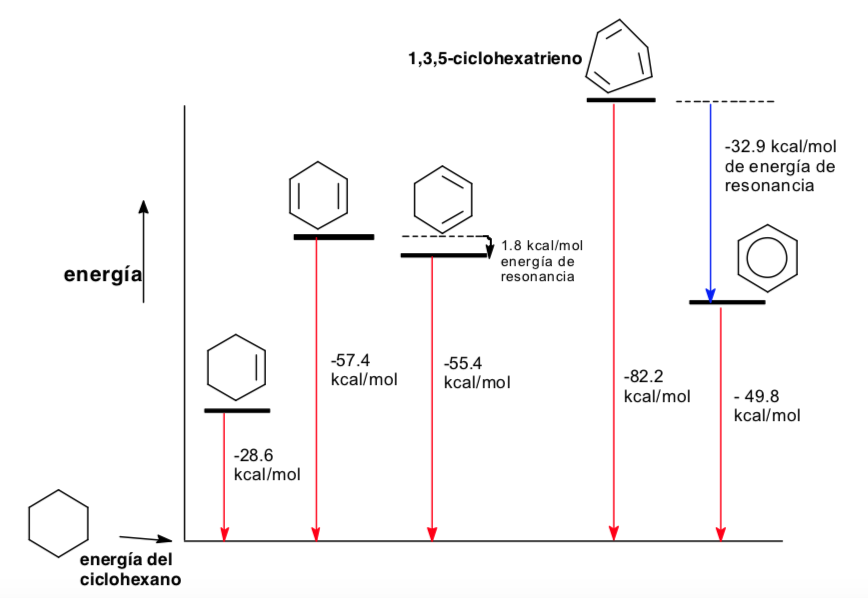 Thermodynamics and conformational analysis (stereoelectronic effects)
Thermodynamics and conformational analysis (stereoelectronic effects)
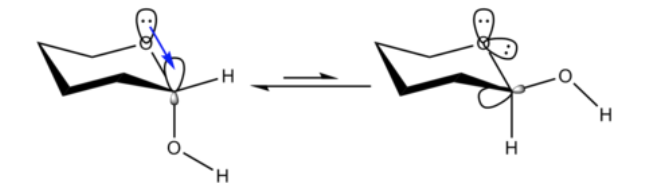
It is not unexpected that the methoxy substituent on a cyclohexane ring prefers to adopt the equatorial conformation.
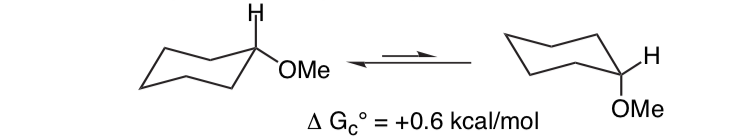
What is unexpected is that the closely related 2-methoxytetrahydropyran ClHO prefers the axial conformation:
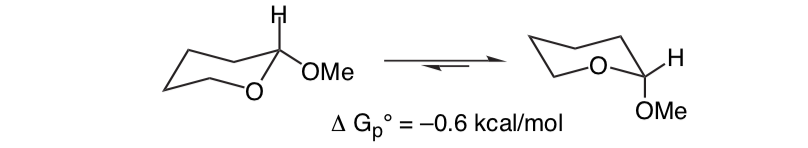
This effect that provides stabilization of the axial O-shaper that abrogates the inherent steric bias of the substituent is called the anomeric effect.
The relationship of free energy with respect to the anomeric effect would be as follows:
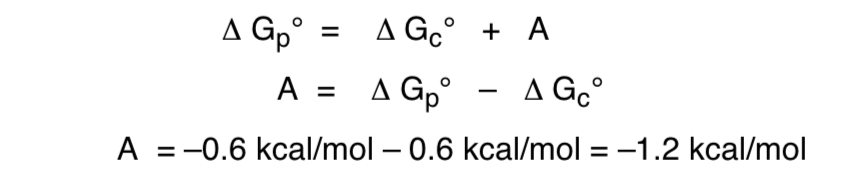
Recommended sources to expand the search:
1) Juaristi E. “Basic concepts of orbital theory”. CINVESTAV, Mexico, 1988 Juaristi E. "Organic Physicochemistry." CINVESTAV, Mexico, 1994.
2) Neil SI “Physical Organic Chemistry” Longman, Milan, 1995.
3) March J., “Advanced Organic Chemistry” John Wiley & Sons, New York, 1992 4) Jones RAY “Physical and Mechanistic Organic Chemistry”, 2nd. Ed Cambridge University Press, Cambridge, 1984.
5) Woodward RB and Hoffmann R. “The conservation of orbital symmetry”, Academic Press, New York, 1979.
6) Carpenter BK “Determination of Organic Reaction Mechanisms”, John Wiley & Sons, New York, 1984 .