Espectroscopia de infrarrojo
La espectrometría del infrarrojo es sumamente útil para determinaciones cualitativas de compuestos orgánicos y para deducir estructuras moleculares a partir de sus grupos funcionales tanto de compuestos orgánicos como inorgánicos.
En el análisis cualitativo la espectroscopia de infrarrojo puede usarse para la identificación de sustancias puras o para la absorción, localización e identificación de impurezas.
Para localizar una impureza en una sustancia se hace una comparación en el espectro de las sustancia que se estudia y una muestra de la sustancia pura. Las impurezas causan bandas de absorción adicionales que aparecen en el espectro.
En el IR también están encontrando uso cada vez mayor en el análisis cuantitativo, el principal campo de aplicación de este tipo de análisis se halla en la cuantificación de contaminantes atmosféricos que provienen de procesos industriales.
Una parte del espectro electromagnético que se extiende desde 0.8 a 1000μm (que corresponde al número de onda comprendidos entre los 12800 y los 10 cm-1), se considera como la región del infrarrojo la cual está dividida en tres regiones llamadas:
a).- I.R. Cercano. b).- I.R. Fundamental ó Medio c).- I.R. Lejano
Cada tipo de enlace absorbe radiación infrarroja a una frecuencia distinta, lo que permite determinar que tipo de grupos funcionales posee la molécula en estudio. Los espectrofotómetros de infrarrojo trabajan en el infrarrojo medio y hacen un barrido desde los 4000 cm−1 hasta los 400 cm−1
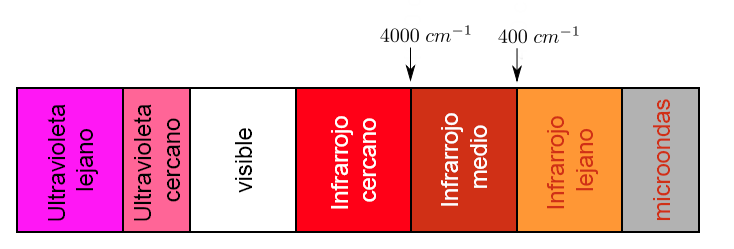
Sin embargo la región de importancia analítica es la región del I.R. Fundamental ya que la mayoría de los instrumentos infrarrojos cubren ésta región.
La mayoría de los materiales orgánicos e inorgánicos demuestran absorción y el espectro es originado principalmente por el alargamiento vibracional y flexión dentro de la molécula. El espectro infrarrojo es una de las propiedades más características de un compuesto ya que no existen dos espectros iguales para dos compuestos diferentes, es como una huella dactilar.
Dentro de la región del I.R. Fundamental existen dos regiones, una de ellas es la llamada de
los grupos funcionales de 4000cm-1 a 1300 cm-1, y la región dactilar de 1300 cm-1 a 670cm-1 .
En la región de los grupos funcionales la posición del pico de absorción es mayor o menor dependiendo solamente del grupo funcional donde llega la absorción y no de la estructura molecular completa. La posición de los picos en la región dactilar son dependientes de la estructura molecular completa.
Características que debe tener una vibración para que produzca banda de absorción:
La radiación incidente debe tener una frecuencia igual a la frecuencia de la vibración que va a producir.
Que la vibración resultante produzca cambio en el momento dipolar, o sea que la vibraciónno absorberá radiación infrarroja, si no hay cambio en el momento dipolar se llama vibración inactiva y serán activas cuando haya dicho cambio en el momento dipolar.
Tradicionalmente, en el eje x de los espectros de infrarrojo se emplea el número de ondas (ν¯, léase "nu barra"') y se define como el inverso de la longitud de onda en cm. ν¯=1λ. En el eje y se representa el porcentaje de radiación transmitida (transmitancia) que se representa por %T. A continuación, se muestra la forma que presenta el espectro de infrarrojos del hexano.
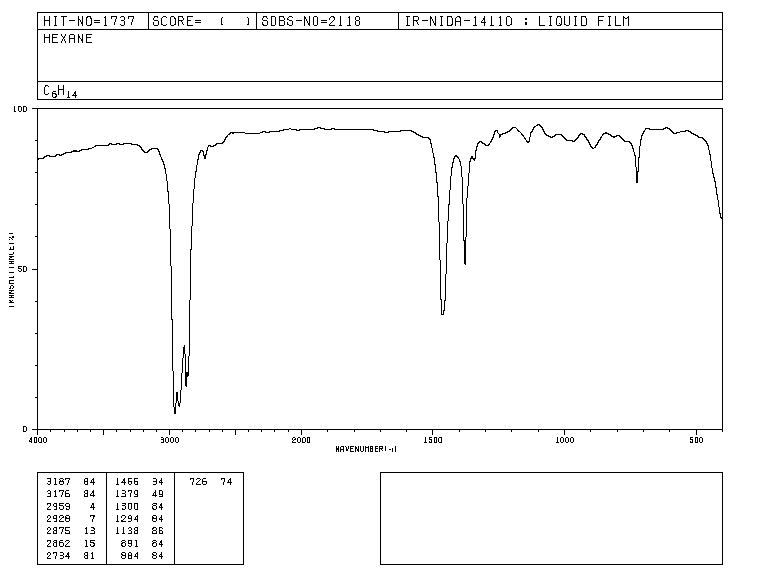
Las bandas representan zonas donde los enlaces de la molécula absorben radiación infrarroja. En las bandas la transmitancia es pequeña y la absorbancia grande.
Tipos de vibracion
Vibración de tensión (stretching). Los átomos unidos por enlaces simples, dobles o triples se acercan y alejan siguiendo la dirección del enlace, igual que oscilan dos masas unidas por un muelle.
Hay dos modos de vibración de tensión: simétrica y asimétrica
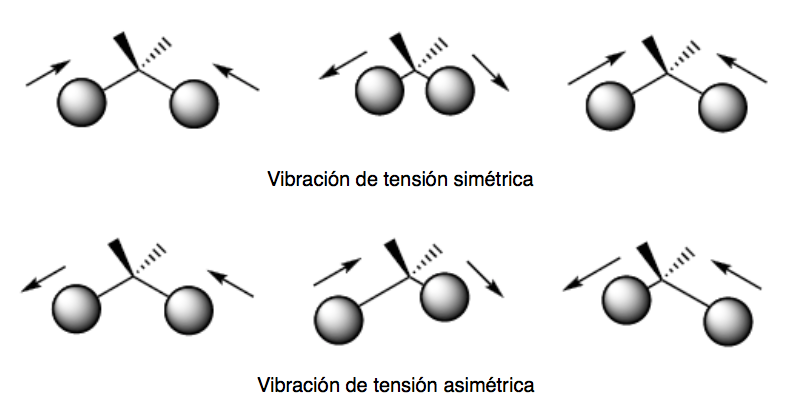
vibración de flexión (bending). Los átomos vibran de modo que varían los ángulos, pero no las longitudes de enlace. Hay cuatro modos de vibraciones de flexión: tijera (scissoring), balanceo (rocking), cabeceo (wagging) y torsión (twisting)
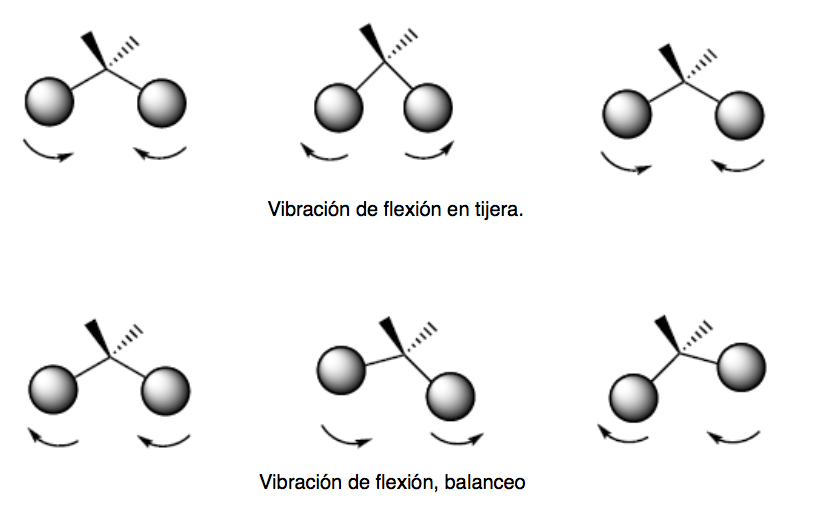
Estos dos modos de vibración tienen lugar en el plano que contiene los tres átomos que participan en la vibración
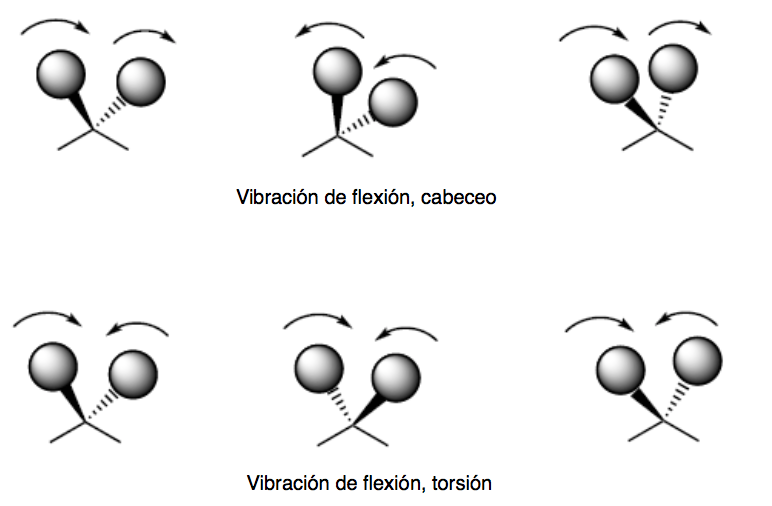
Los modos de vibración de cabeceo y torsión tienen lugar fuera del plano (Out of plane) y suelen representarse por Oop.
Oscilador armonico cuantico
Las vibraciones moleculares pueden estudiarse con el modelo del oscilador armónico cuántico. La energía viene dada por:
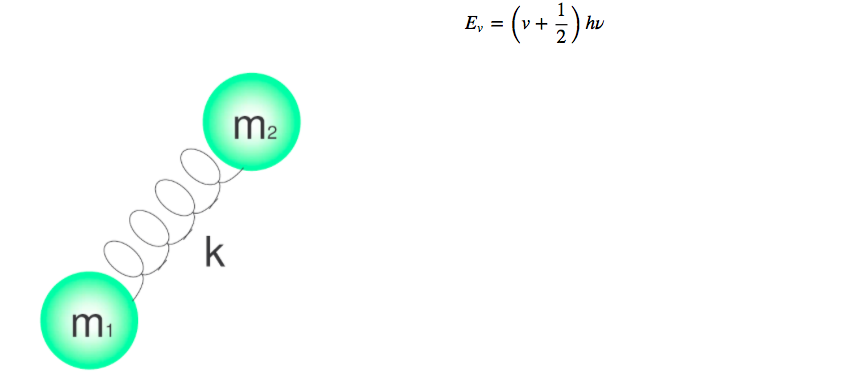
Los distintos niveles de energía vienen dados por el número cuántico v, que toma valores 0.1.2.3.4.....
h es la constante de Planck y ν la frecuencia del oscilador que viene dada por la expresión:
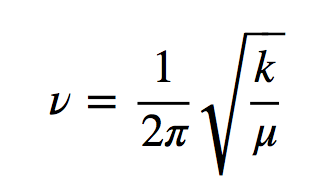
Donde k es la constante de fuerza del muelle y μ la masa reducida del sistema
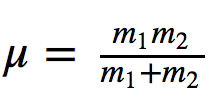
Dividiendo la frecuencia entre la velocidad de la luz se obtiene número de ondas ν¯
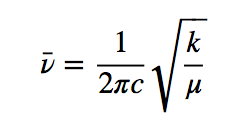
La ecuación anterior indica que masas reducidas pequeñas (átomos de poca masa) y constantes de fuerza altas (enlaces fuertes) conducen a frecuencias altas. En estas condicionees las bandas de absorción salen a numeros de onda altos.
Como puede observarse en el gráfico las frecuencias altas dan lugar a un mayor espaciado entre los niveles energéticos.
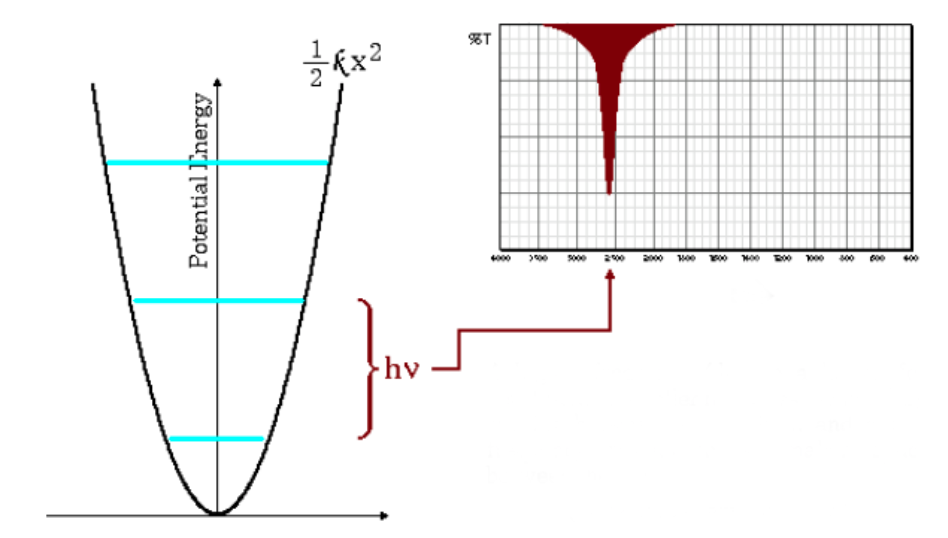
Frecuencias de absorción bajas
La ecuación antetior tambien indica que masas reducidas grandes y constantes de fuerza pequeñas (enlaces débiles) conducen a frecuencias bajas. En estas condicionees las bandas de absorción salen a numeros de onda bajos.
Como puede observarse en el gráfico las frecuencias bajas dan lugar a un menor espaciado entre los niveles energéticos.
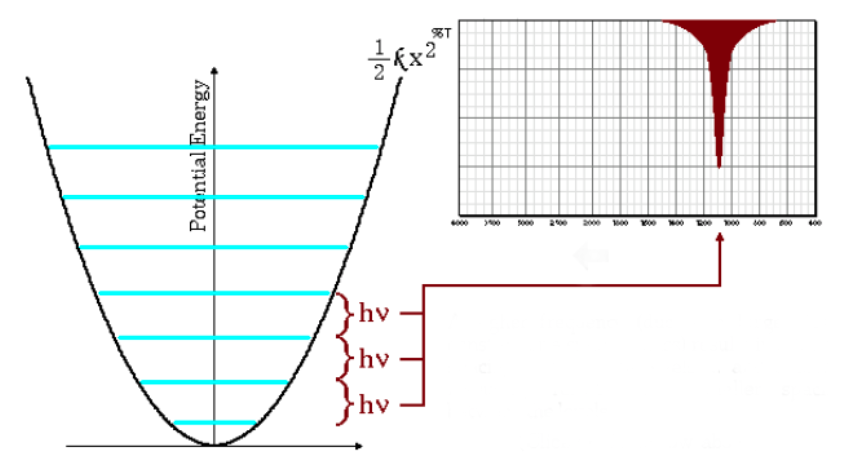
Principales vibraciones moleculares
Espectro IR
En un espectro de infrarrojos se representa la frecuencia (en número de onda) frente al porcentaje de luz transmitida (transmitancia). El porcentaje de transmitancia se define como el cociente entre la intensidad de la luz transmitida a través de la muestra, IM, y la intensidad de la luz del haz de referencia IR multiplicado por 100.
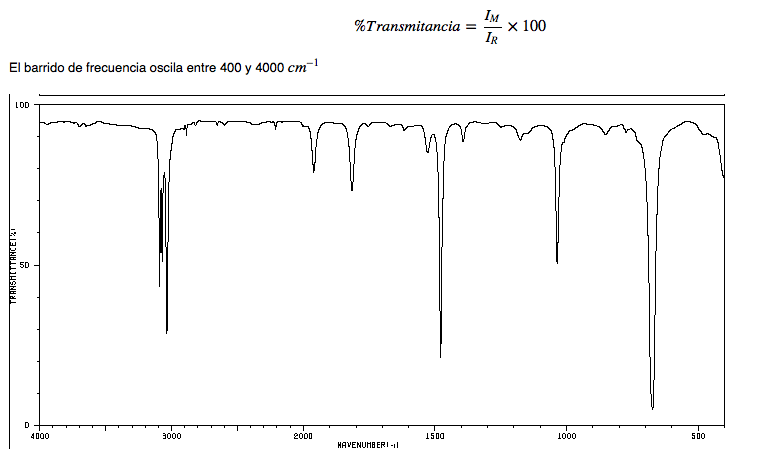
Espectro IR de alcanos
• Tensión C-H: los alcanos presentan vibraciones de tensión C-H ligeramente por debajo de 3000 cm−1
• Flexión C-H: los CH2 de la cadena presentan vibraciones de flexión (tijera) a 1465 cm−1, mientras que los metilos producen una banda a 1375 cm−1debida a la vibración de flexión simétrica y otra a 1450 cm−1 debida a la vibración de flexión asimétrica. Todas las bandas de flexión son de intensidad media.
Obsérvese que la banda de flexión asimétrica del metilo solapa con la de flexión en tijera del CH2.
Espectro IR del hexano
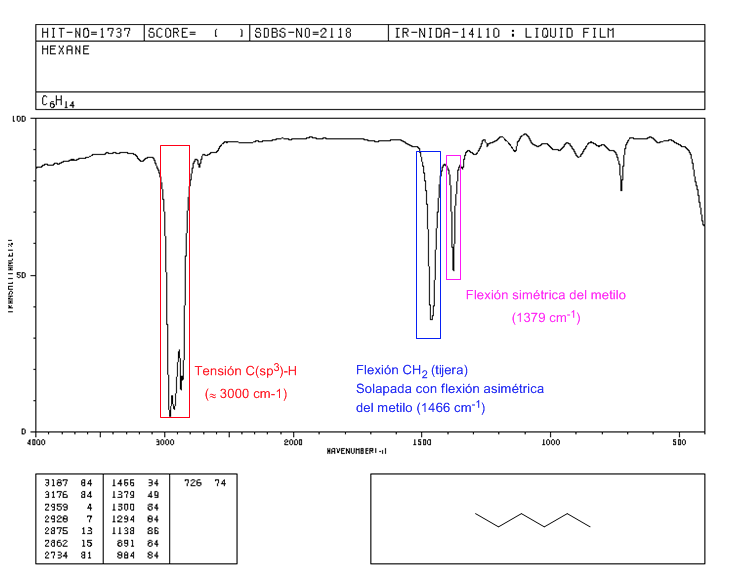
Espectro del 2,2-dimetilbutano
La presencia del grupo tert-butilo produce el desdoblamiento de la banda de flexión simétrica en dos bandas a 1390 y 1370 cm−1. La banda a 1390 tiene la mitad de intensidad que la de 1370.
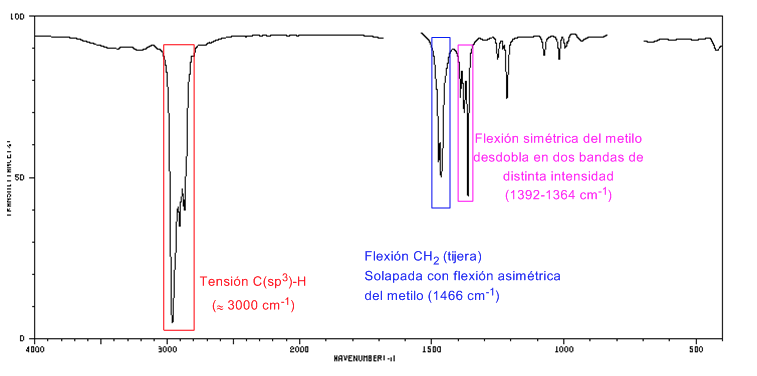
Espectro IR de cicloalcanos
Los cicloalcanos tienen un espectro de IR muy similar a los alcanos con banda de tensión C-H ligeramente por debajo de 3000 cm−1 y banda de flexión C-H en tijera para los CH2 a 1465 cm−1. La principal diferencia con los alcanos es la ausencia de la banda de tensión simétrica del metilo.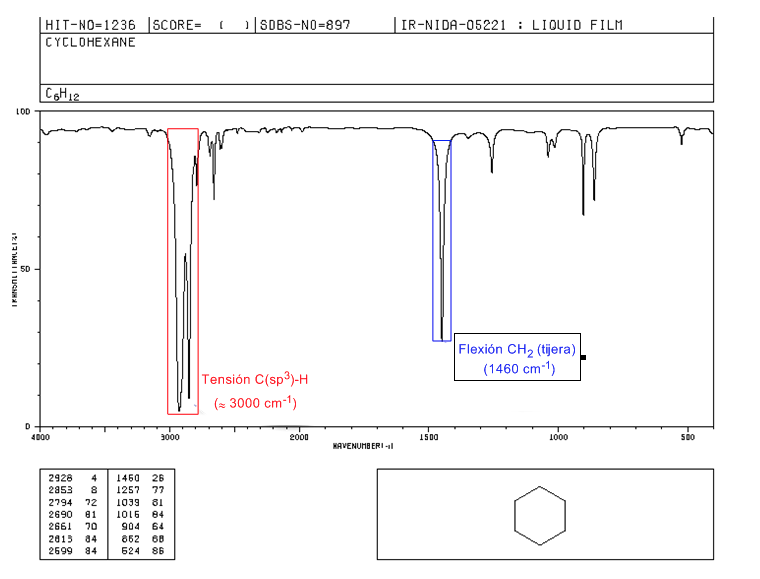
Obsérvese la ausencia de la banda de flexión simétrica del metilo que presentan los alcanos a 1375 cm−1.
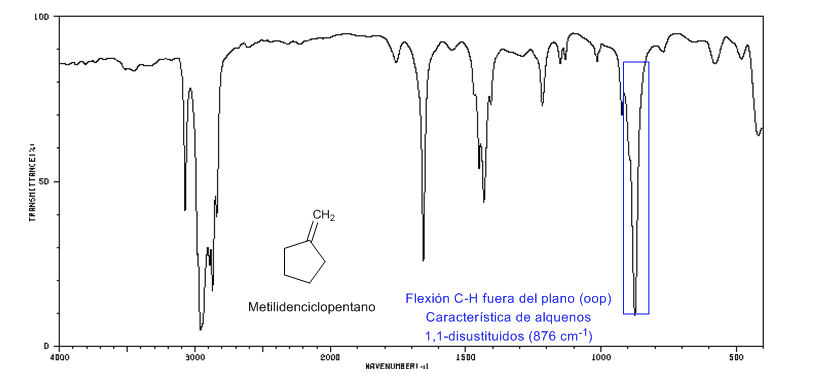
Espectro IR de alquenos
• Tensión C(sp2)-H: 3100 -3000 cm-1
• Tensión C=C: 1600 cm-1
• Flexión fuera del plano (oop) del enlace C=C-H: 1000 - 650 cm-1. Este tipo de banda permite conocer el grado de sustitución del alqueno.
Espectro del 1-penteno
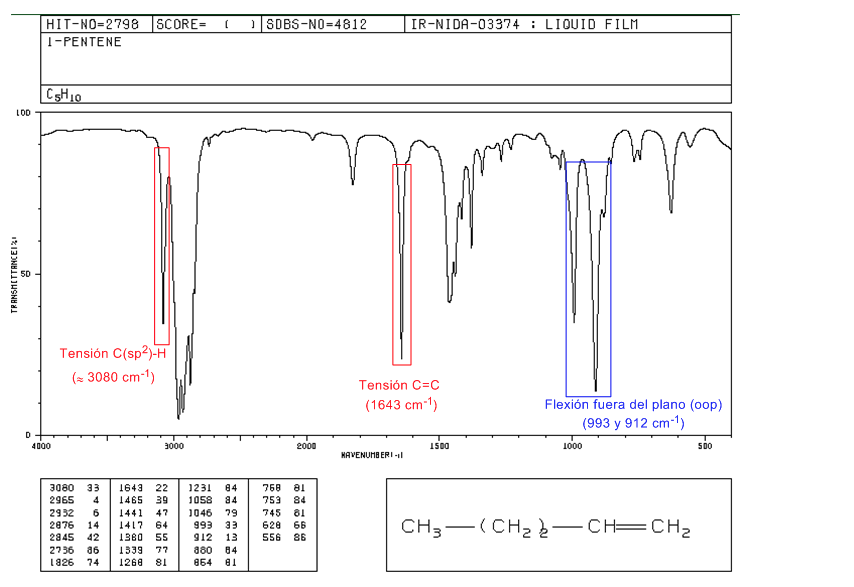
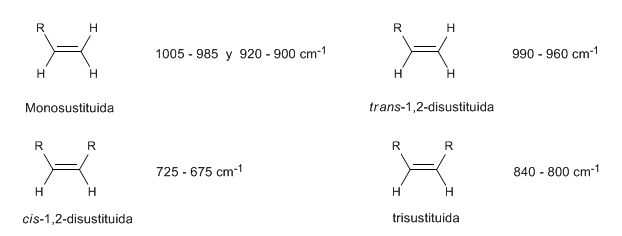
En los alquenos monosustituidos, como el 1-penteno, las flexiones C-H fuera del plano producen dos bandas situadas en 1005-985 y 920-900 cm−1.
Estereoquimica y espectroscopia IR de alquenos
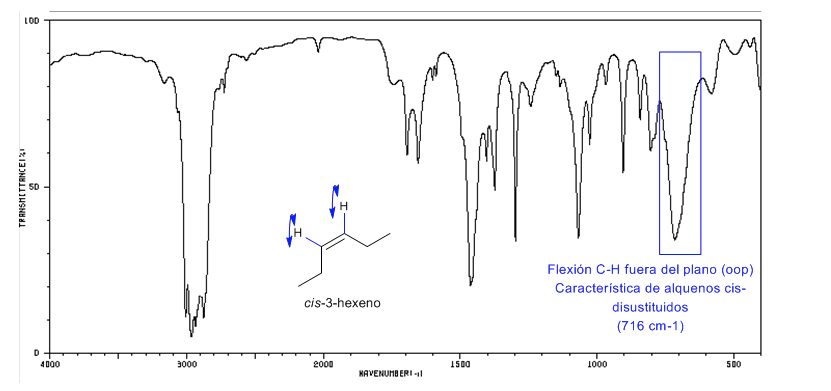
Los alquenos cis-disustituidos presenan una banda de flexión C-H fuera del plano que permite distinguirlos. Esta banda aparece entre 725-675 cm−1
Espectro del Cis-3-hexeno
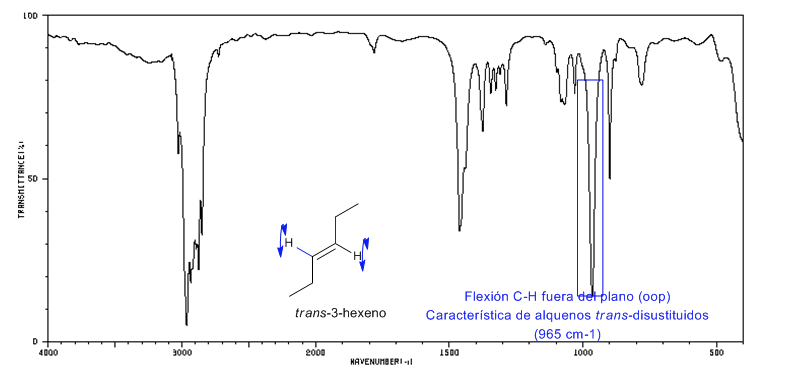
Espectro del Trans-3-hexeno
Los alquenos trans-disustituidos presentan una banda de absorción fuerte entre 980-965 cm−1 que permite identificarlos. Obsérvese la ausencia total de la banda de tensión C=C a 1600 cm−1 debido a la falta de polaridad.
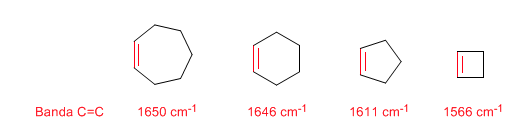
Tensión de anillo: enlaces C=C endocíclicos
Al disminuir el tamaño del anillo la banda de tensión de los enlaces C=C se desplaza hacia menor número de ondas. La excepción del ciclopropeno se atribuye al acoplamiento entre las vibraciones de tensión de los enlaces C=C y C-C. Este acoplamiento no se produce en el ciclobuteno debido a que los enlaces C=C y C-C se encuentran perpendiculares entre sí.
 Espectro IR del ciclopenteno
Espectro IR del ciclopenteno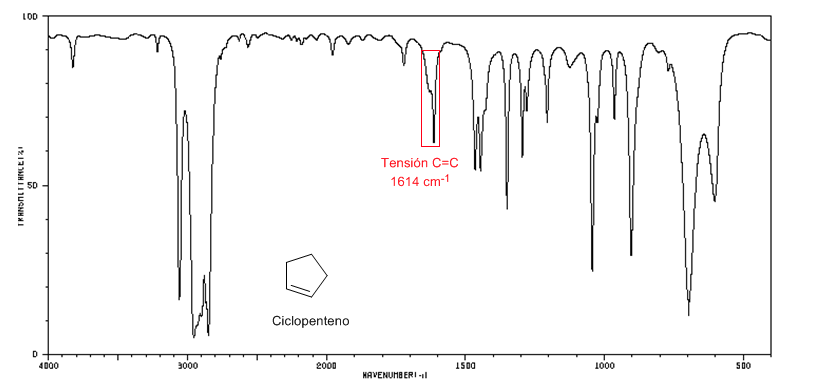
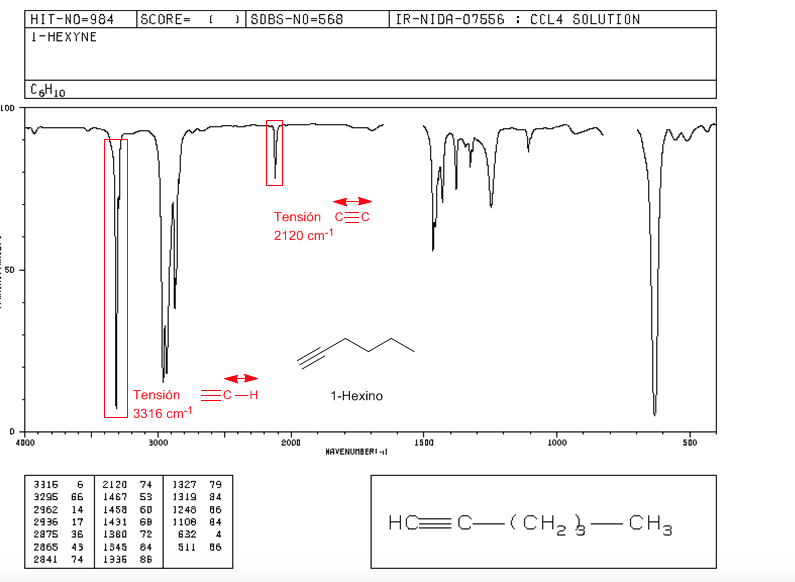
Tensión ≡C−H: 3300 cm−1
Tensión −C≡C−: 2150 cm−1. Los alquinos simétricos no presentan esta banda, siendo muy débil en los internos. La conjugación baja ligeramente el valor.
Espectro del 1-hexino
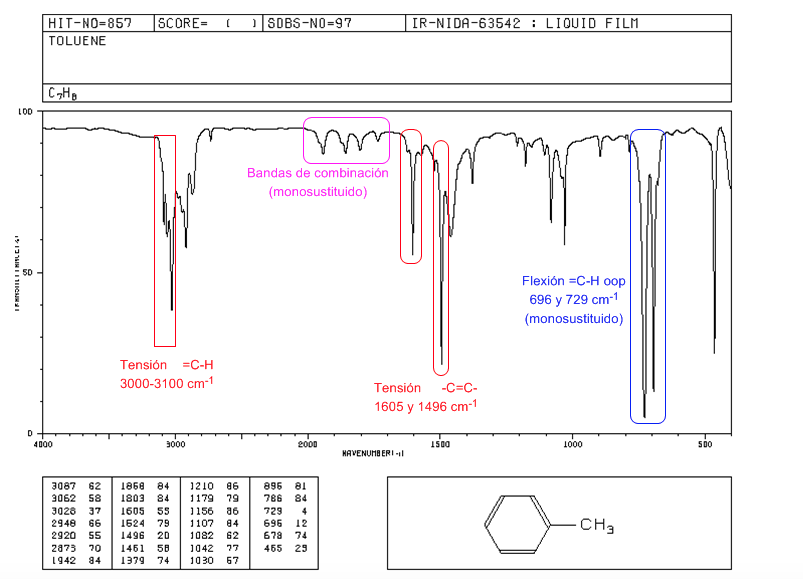
Espectro IR de aromaticos
Tensión =C-H: 3100 cm-1
Tensión -C=C-: 1600 y 1475 cm-1
Flexión =C-H fuera del plano: 900-690 cm-1
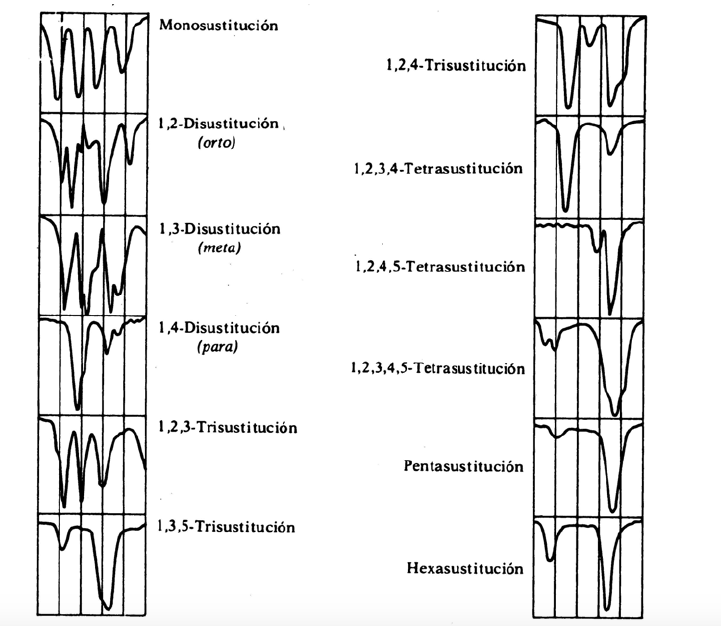
Las vibraciones oop junto con los sobretonos y bandas de combinación que aparecen entre 2000 y 1667 cm-1 permiten conocer el grado de sustitución del benceno.
Bandas de sustitucion
![]()
Espectros de alcoholes y fenoles
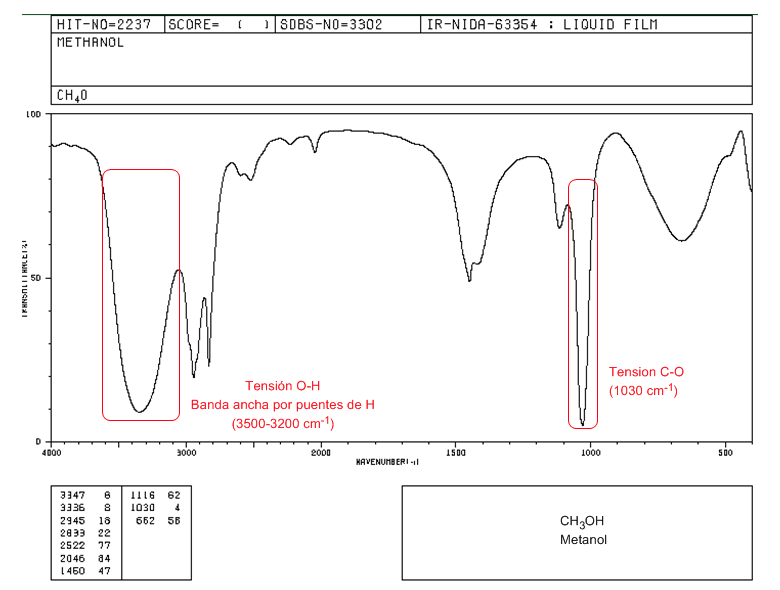
Tensión O-H: Banda ancha desde 3500 a 3200 cm-1. En ausencia de puentes de hidrógeno aparece como un pico agudo a 3650-3600 cm-1.
Tensión C-O: Banda comprendida entre 1250-1000 cm-1. Permite distinguir entre alcoholes primarios (1050 cm-1), secundarios (1100 cm-1), terciarios (1150 cm-1) y fenoles (1220 cm-1).
Espectro IR de un alcohol primario (metanol)
En el espectro del metanol podemos observar la banda de tensión O-H, muy ancha, por formación de puentes de hidrógeno. La banda de tensión C-O sale a número de ondas bajo (1030) por tratarse de un alcohol sin sustituyentes.
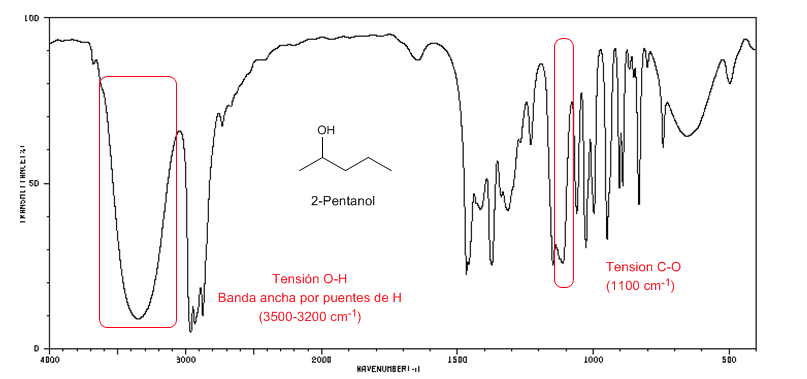
Espectro IR de alcoholes secundarios (2-pentanol)
Obsérvese el desplazamiento de la banda C-O hacia mayor número de ondas con respecto al metanol.
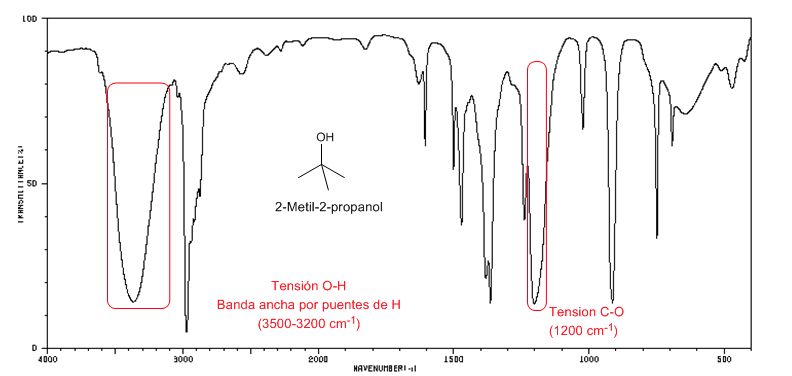
 Espectro IR de alcoholes terciarios ( 2-metil-2-propanol)
Espectro IR de alcoholes terciarios ( 2-metil-2-propanol)Los alcoholes terciarios tienen la banda C-O desplazada a frecuencias mayores que los alcoholes primarios y secundarios.
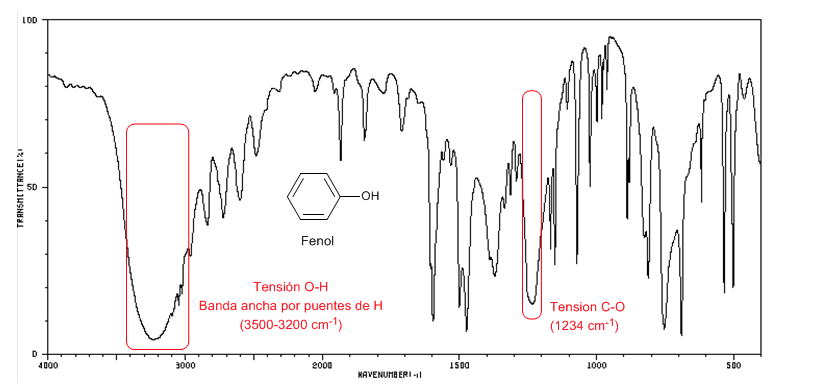
El fenol presenta una banda de absorción C-O por encima de 1200 cm-1
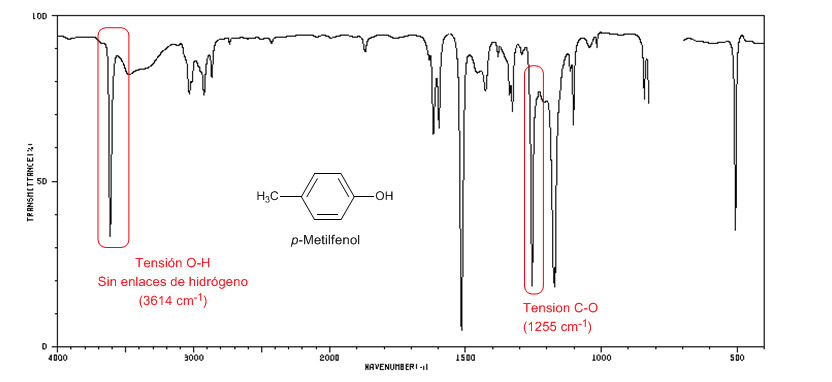
El siguiente espectro muestra la banda de tensión O-H en ausencia de puentes de hidrógeno.
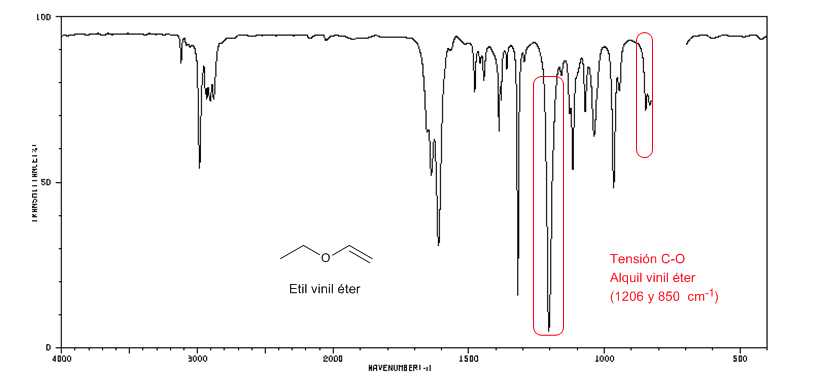
Los alquil vinil éteres (CH2=CH-O-R) presentan dos bandas a 1220 y 850 cm-1. Esta última muy débil.
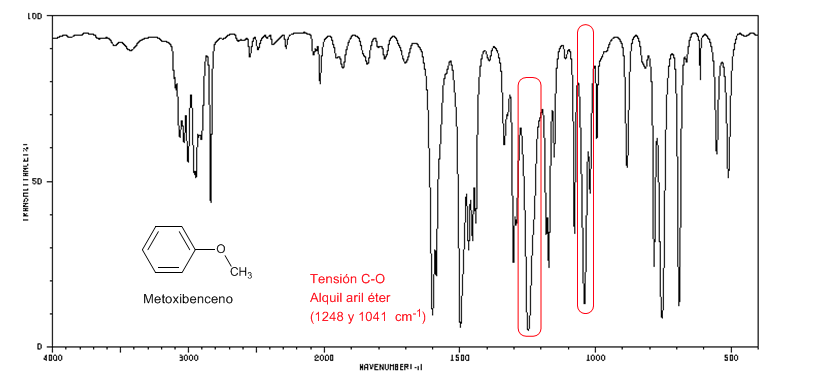
Los aril alquil éteres (Ar-O-R) presentan dos bandas a 1250 y 1040 cm-1
Espectro IR del 1metoxihexano
El etil vinil éter presenta dos bandas a 1220 y 850 cm-1.Esta última muy débil.
El metoxibenceno presenta dos bandas a 1250 y 1040 cm-1
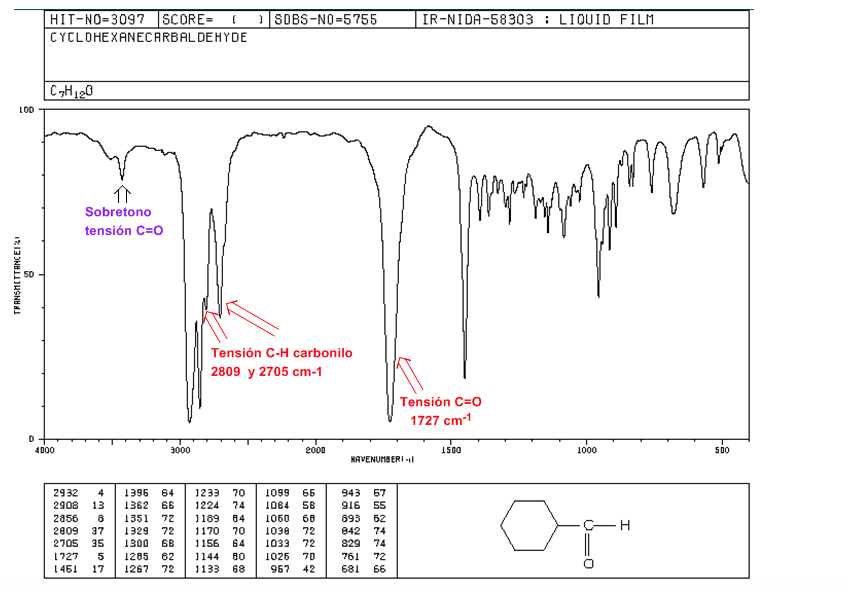
Aldehidos
Tensión C=O: 1725 cm-1
Tensión C-H carbonilo: dos bandas débiles a 2850 y 2750 cm-1. La banda a 2850 suele solaparse con la de tensión C(sp3)-H
Sobretono de Tensión C=O sobre 3500 cm-1.
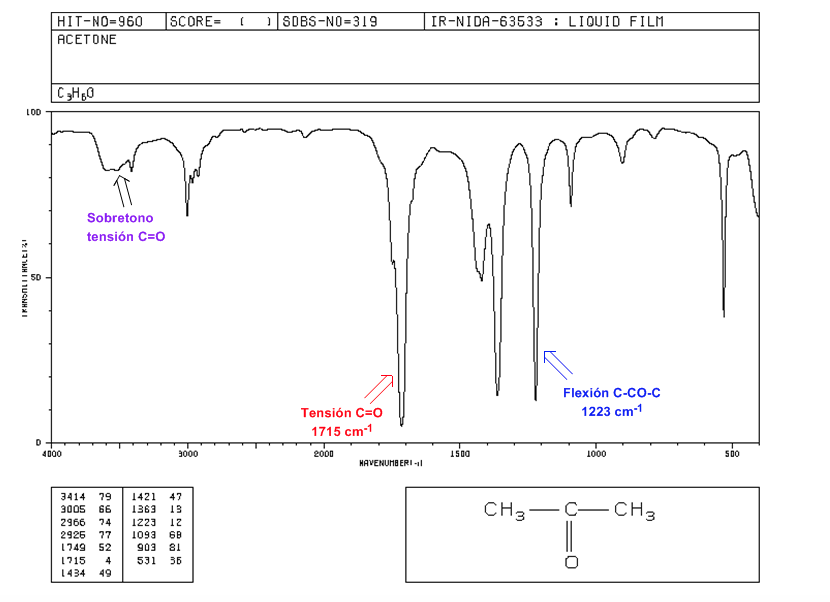
Tensión C=O: Banda intensa a 1715 cm-1.
Flexión C-CO-C: 1300 - 1100 cm-1.
Sobretono de tensión C=O: desde 3500 a 3350 cm-1.
Efecto de la conjugación en la banda de Tensión C=O
Espectro IR de acidos carboxilicos y derivados
Acidos carboxilicos
Tensión O-H: Desde 3400 a 2400 cm-1. Muy ancha debido a la formación de puentes de hidrógeno.
Tensión C=O: 1730-1700 cm-1
Tensión C-O: 1320-1200 cm-1
Flexión C-O-H (oop): Banda en forma de campana a 900 cm-1
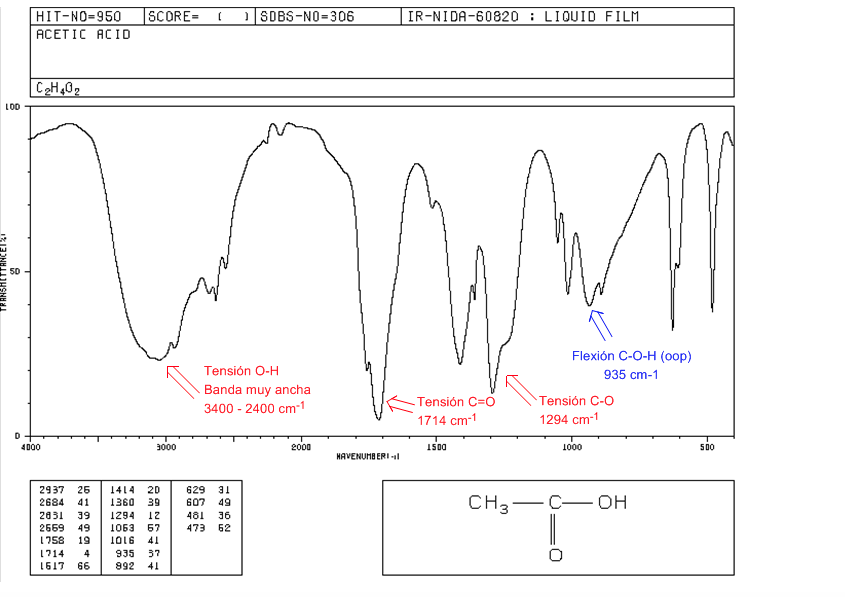
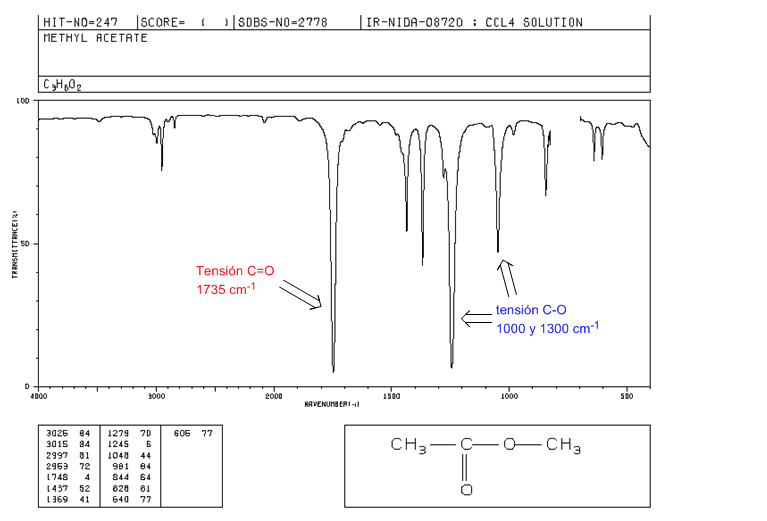
Esteres
Tensión C=O a 1735 cm-1. Si existen dobles enlaces conjugados con el carbonilo la banda se desplaza a valores más bajos. Cuando el doble enlace se encuentra sobre el grupo alcoxi (-OR) del éster se observa un desplazamiento hacia valores más altos.
Tensión C-O: 2 bandas a 1300 y 1000 cm-1. Siendo más ancha e intensa la obsevada a 1300.
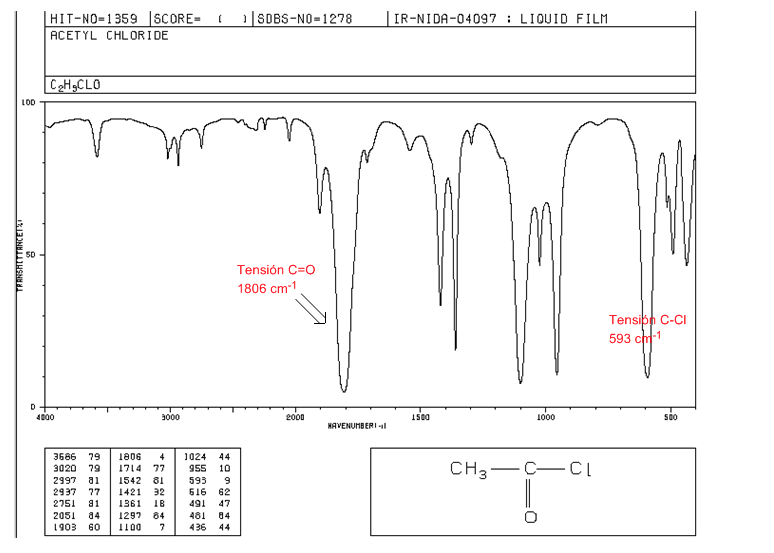
Haluros de acido
Tensión C=O: 1810 - 1775 cm-1
Tensión C-Cl: banda intensa 730 - 550 cm-1
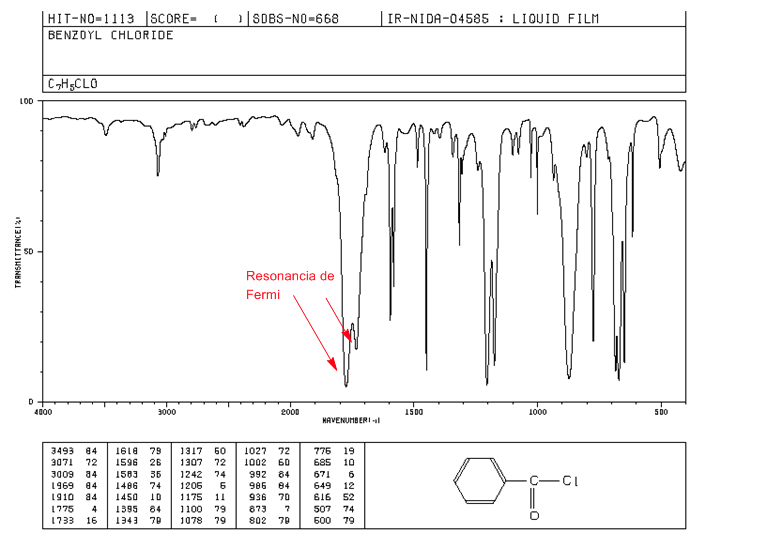
Resonancia de Fermi
Sobretonos, son transiciones vibracionales desde el estado fundamental a estados excitados superiores. Las frecuencias de absorción son siendo la frecuencia de la absorción fundamental. Resonancia de Fermi, resultan del acoplamiento de una banda de absorción fundamental con un sobretono o una banda de combinación.
Los haluros de alcanoílo aromáticos presentan dos bandas de tensión C=O por resonancia de Fermi.
 Nitrilos
Nitrilos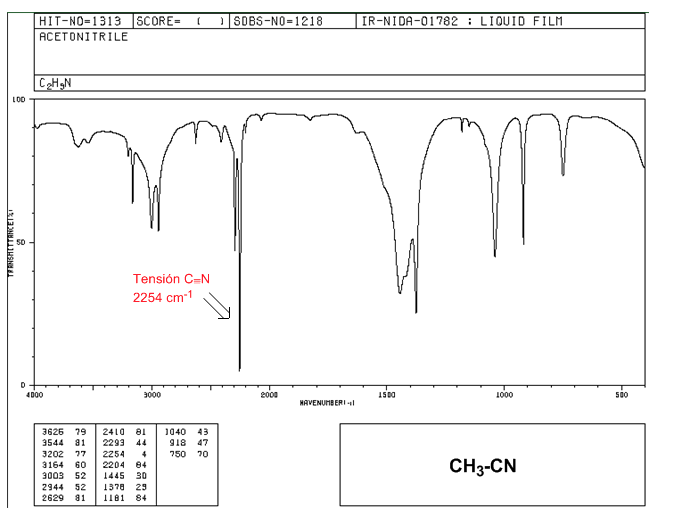
Amidas
Tensión C=O: 1680 - 1630 cm-1
Tensión N-H: Entre 3350 y 3180 cm-1. Las amidas primarias presentan dos bandas, mientras que las secundarias tienen una sóla banda.
Flexión N-H: 1640 - 1550 cm-1
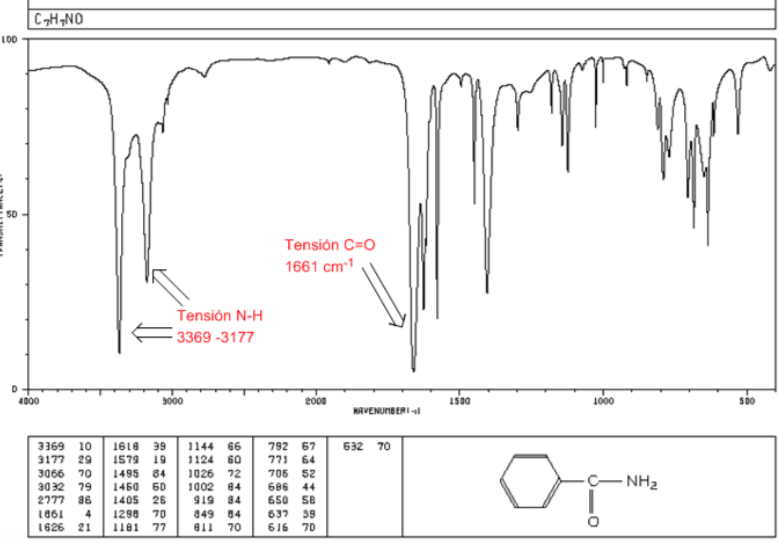
Espectro IR de aminas
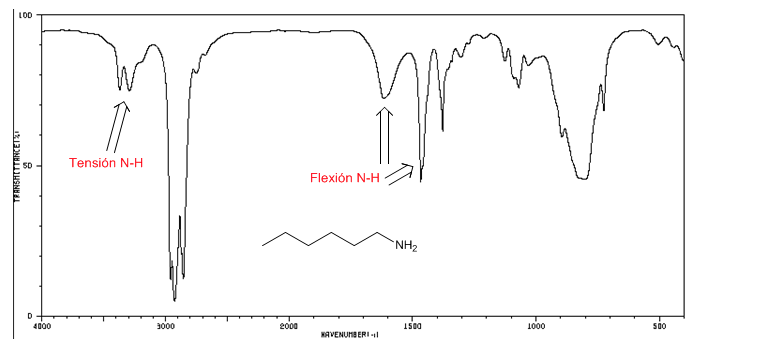
Tensión N-H: entre 3500 y 3300 cm-1. Las aminas primarias presentan dos bandas (simétrica y asimétrica), las secundarias una sóla banda.
Flexión N-H: Aminas primarias dos bandas a 1640 y 1560 cm-1. Secundarias una banda a 1500 cm-1
Espectro IR de la 1-hexano-amina
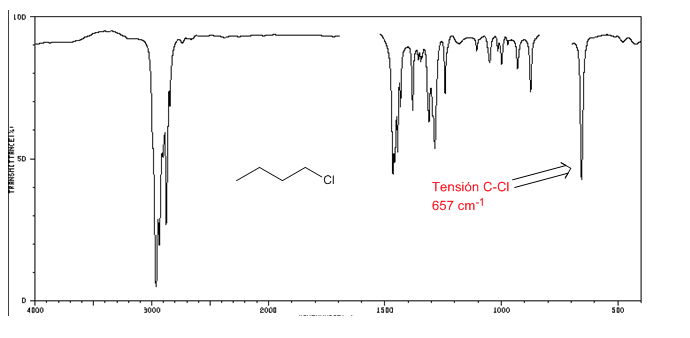
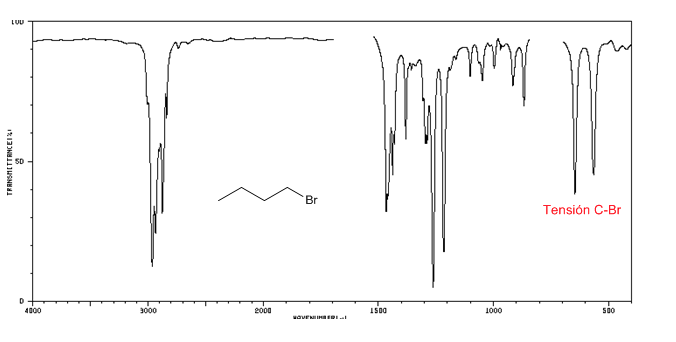
Espectro IR de halogenuros de alquilo
Tensión C-Cl: 785 - 540 cm-1
ensión C-Br: 650 - 510 cm-1
 Fuentes recomendadas para expandir la busqueda:
Fuentes recomendadas para expandir la busqueda:
1. Aga Fano S.A. Espectroscopia de Emisión. (acceso 10 de septiembre del 2007).http://hiq.aga.com.co/International/Web/LG/CO/likelgspgco.nsf/DocByAlias/anal_icp.
2. Alonso, P. et al. quimicaCou..Ed. Mc Graw-Hill. 1990.
3. Álvarez Jiménez, M. D. y Gómez del Río, M. I. Guía didacticaQuímica Analítica II. UNED. 1999.
4. Arribas Jimeno Siro; Burriel Barcelo Fernando; Hernández Méndez Jesús; Lucena Conde Felipe. Química Analítica Cualitativa. ISBN: 8497321405. ISB. 2006.
5. Ayres, Gilbert H. Análisis Químico Cuantitativo. Ediciones del Castillo, 4ta ed. ISBN: 8421902806. 1981.
6. Bermejo Barrera. M del Pilar. Química analítica general, cuantitativa e instrumental. Editorial Paraninfo. 7ma Edición. ISBN: 8428318093. 1990.
7. Blanco, M., Cerdá, V. y Sanz Medel, A., Espectroscopia Atómica Analítica, Publicaciones de la universidadAutónoma de Barcelona. 1990.
8. Brode. R.W, Chemical spectroscopy, Nueva York 1952.
9. Burriel, M.F., Lucena, C.F.Química Analítica Cuantitativa. Edición Revolucionaria. La Habana.1978.
10. Burriel, F. Química Analítica Cualitativa. Editorial Paraninfo. ISBN: 8497321405. pp 1072. , 2003.