
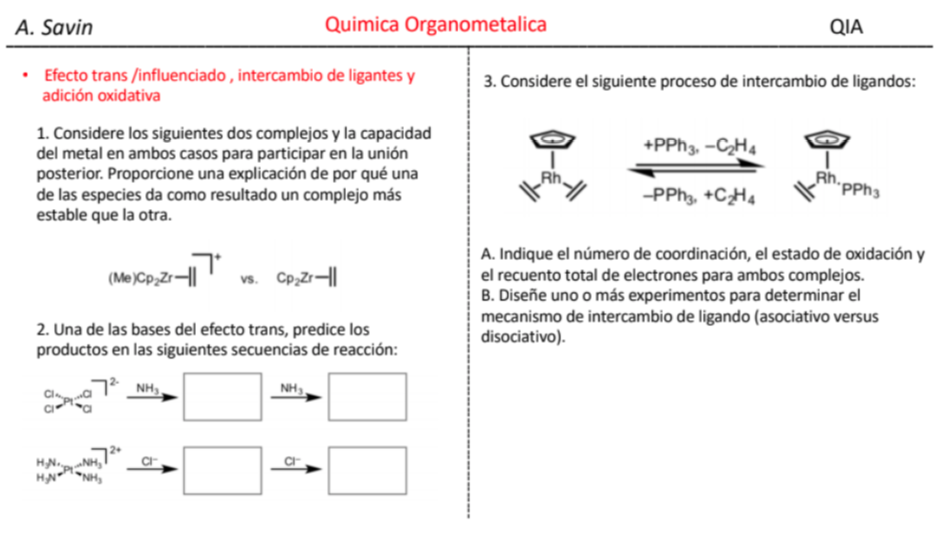
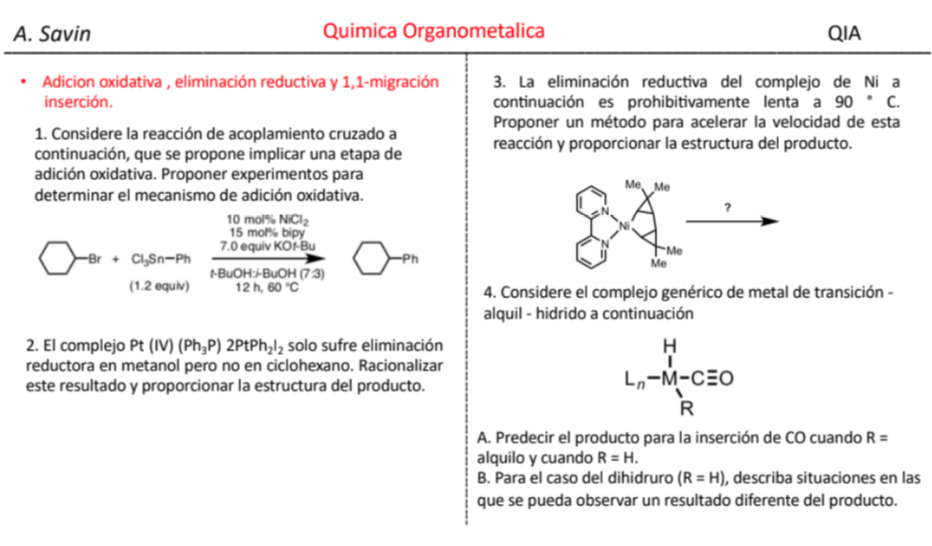
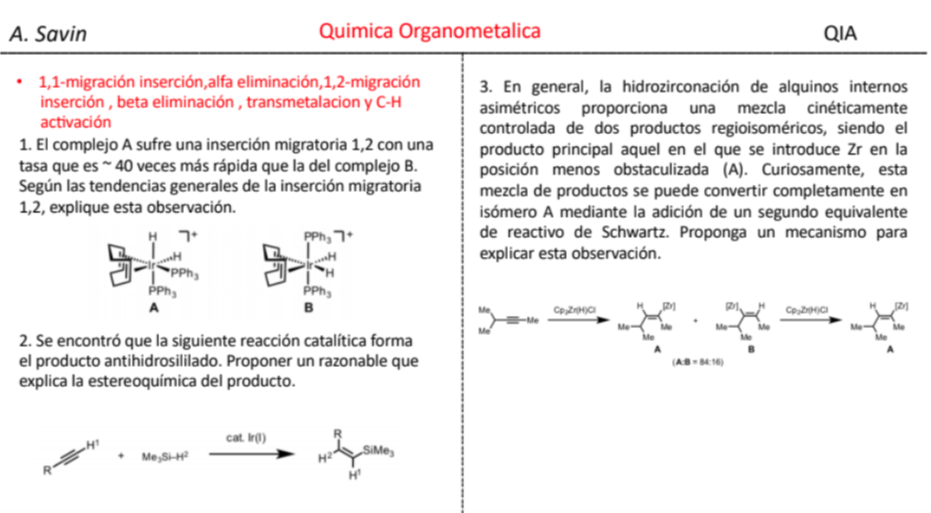
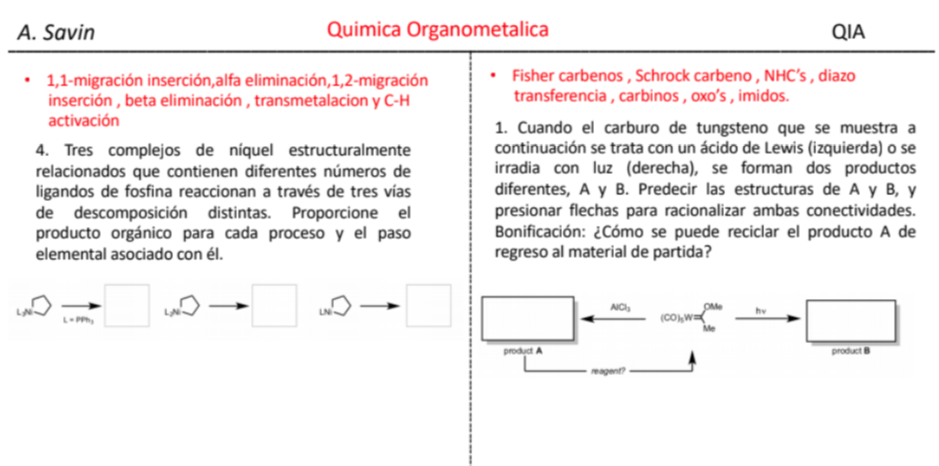


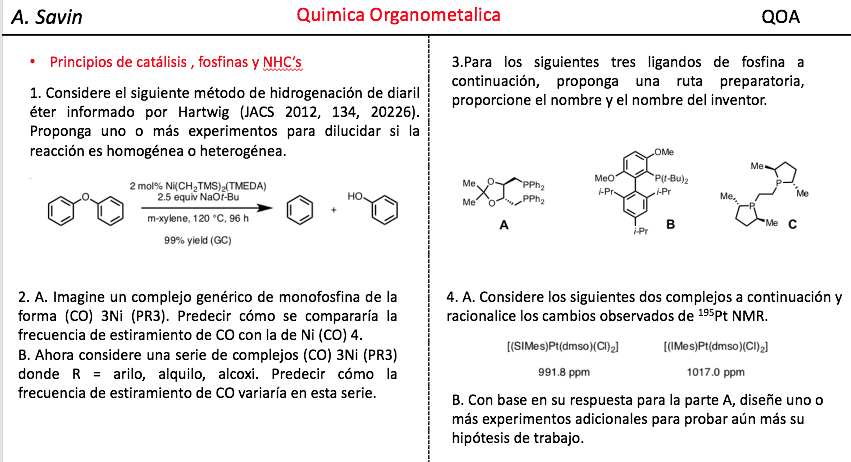
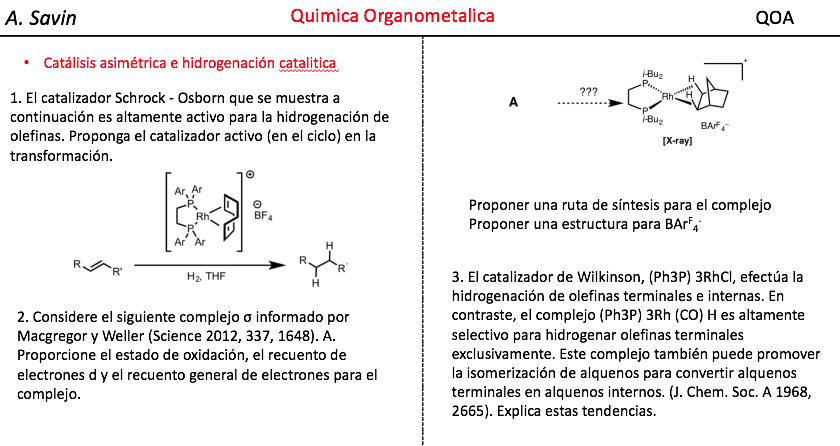
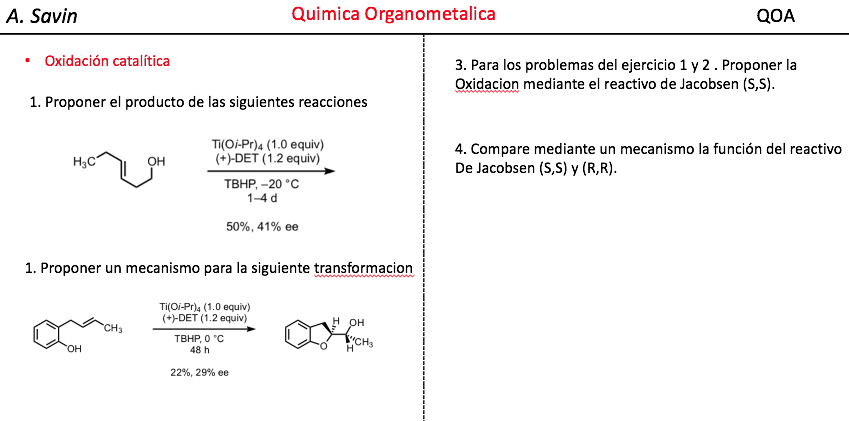
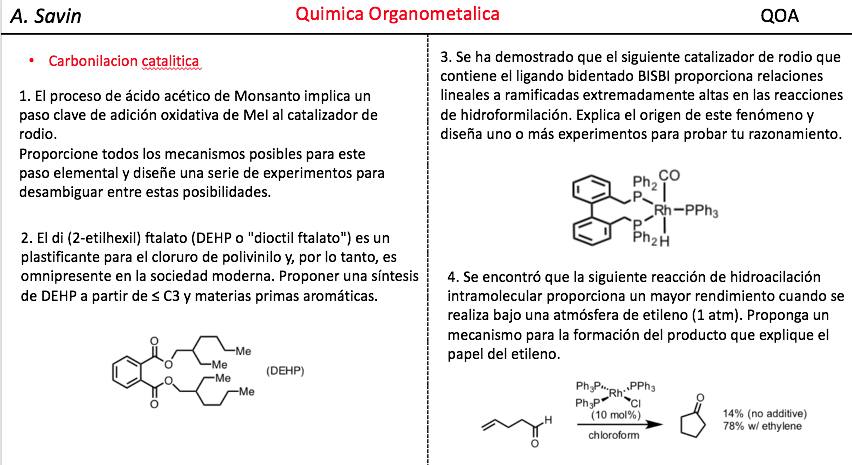
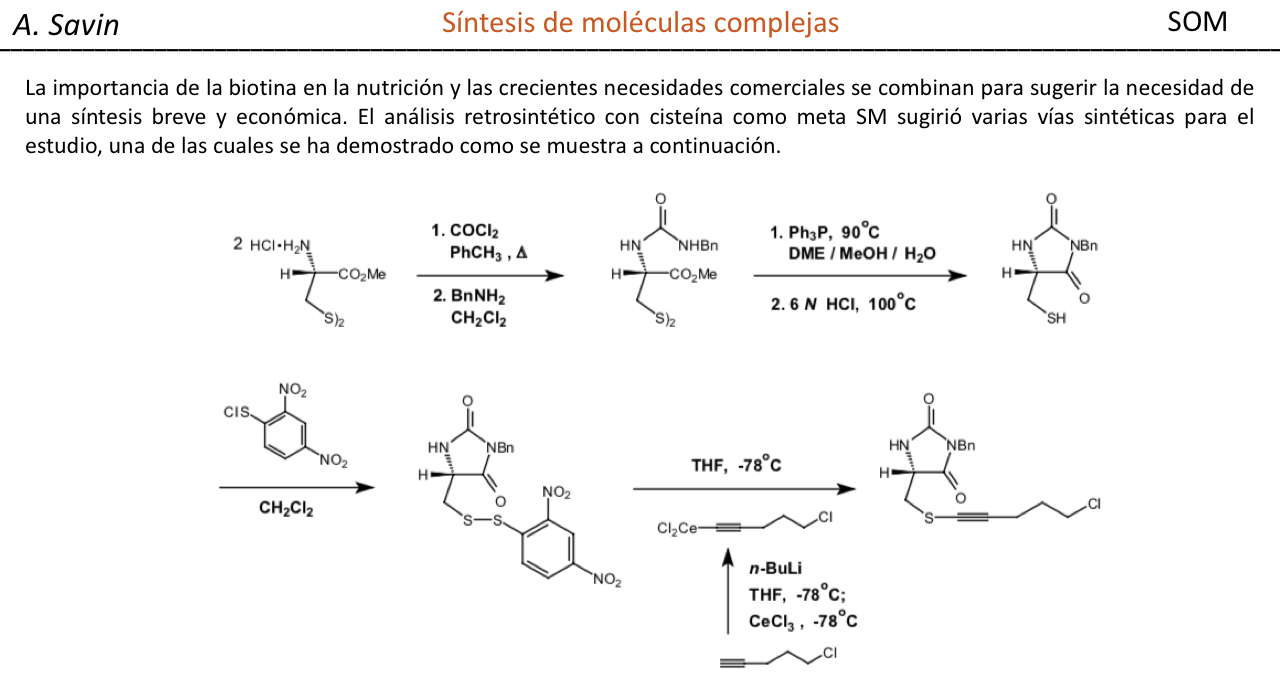
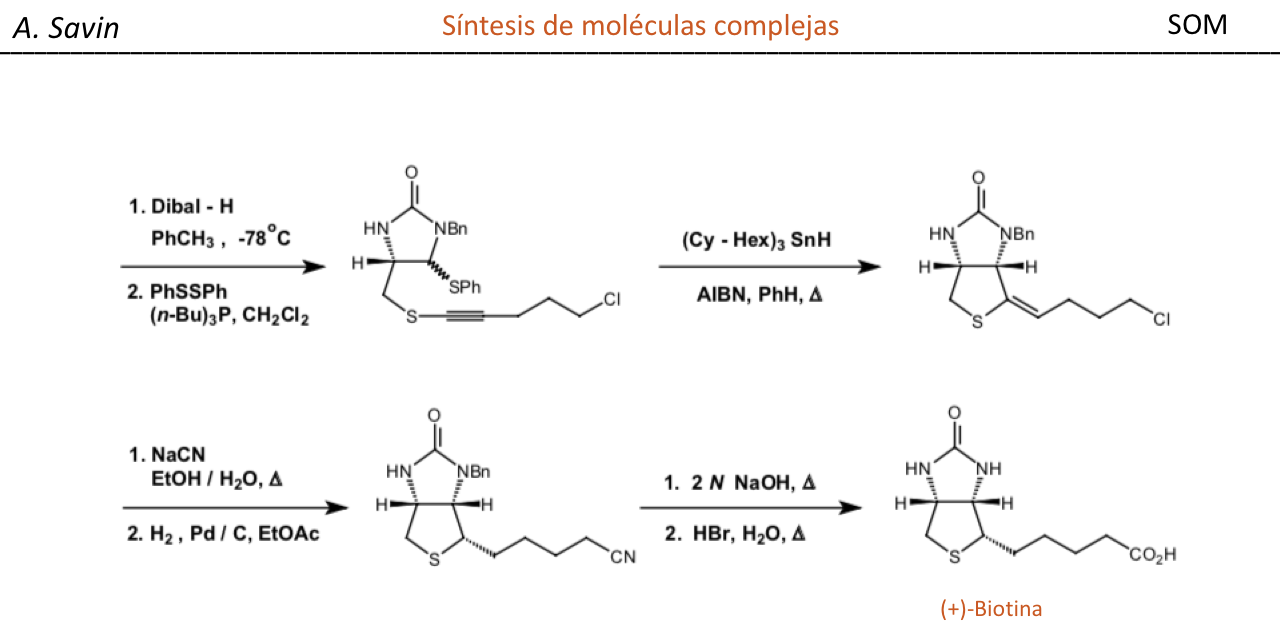
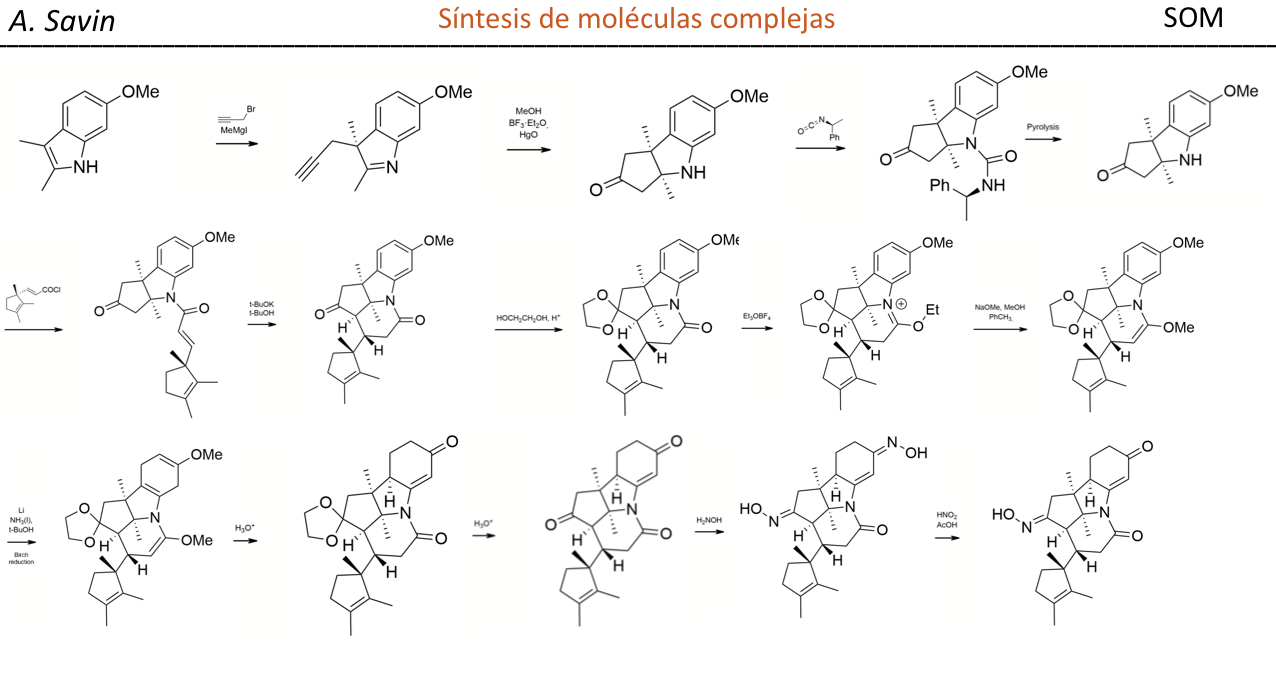
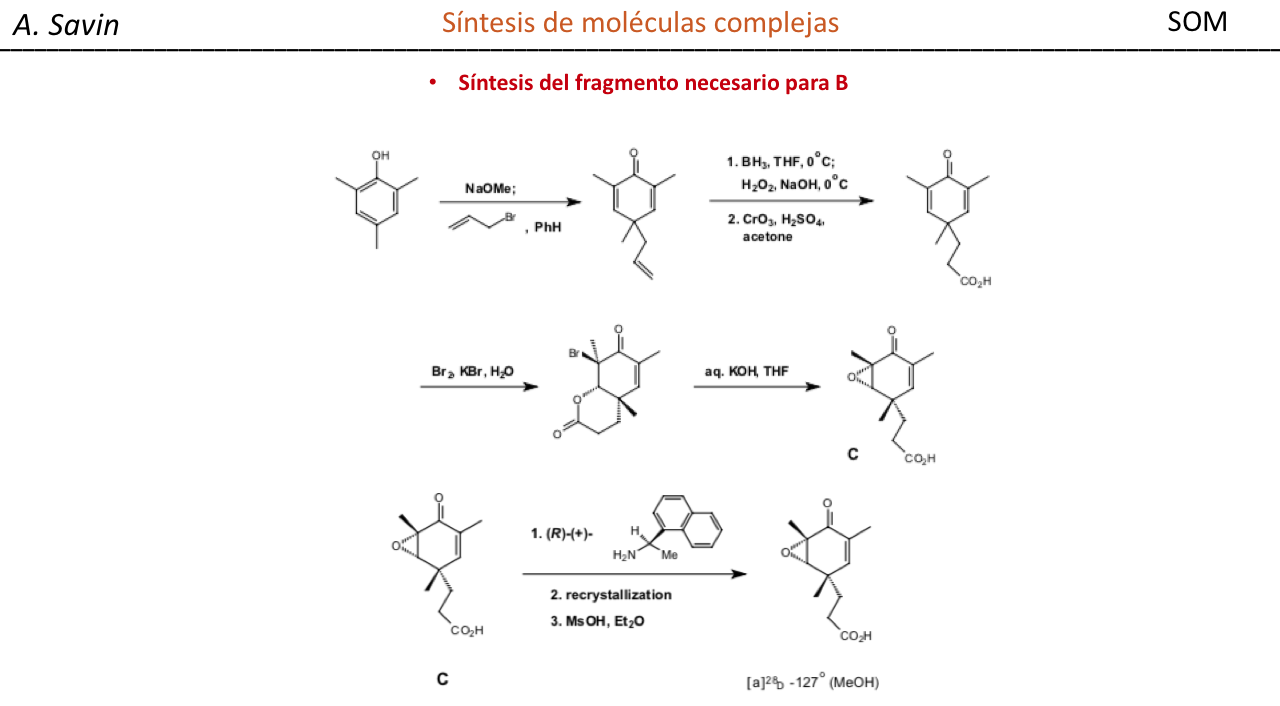
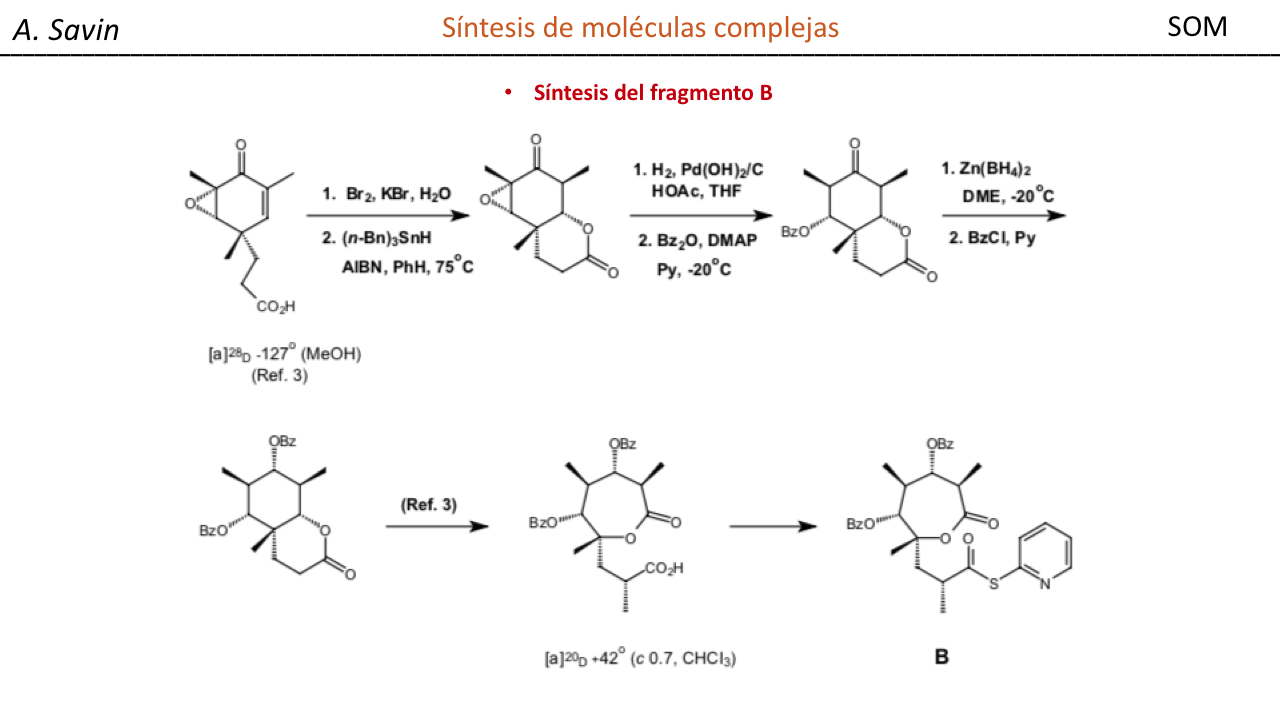
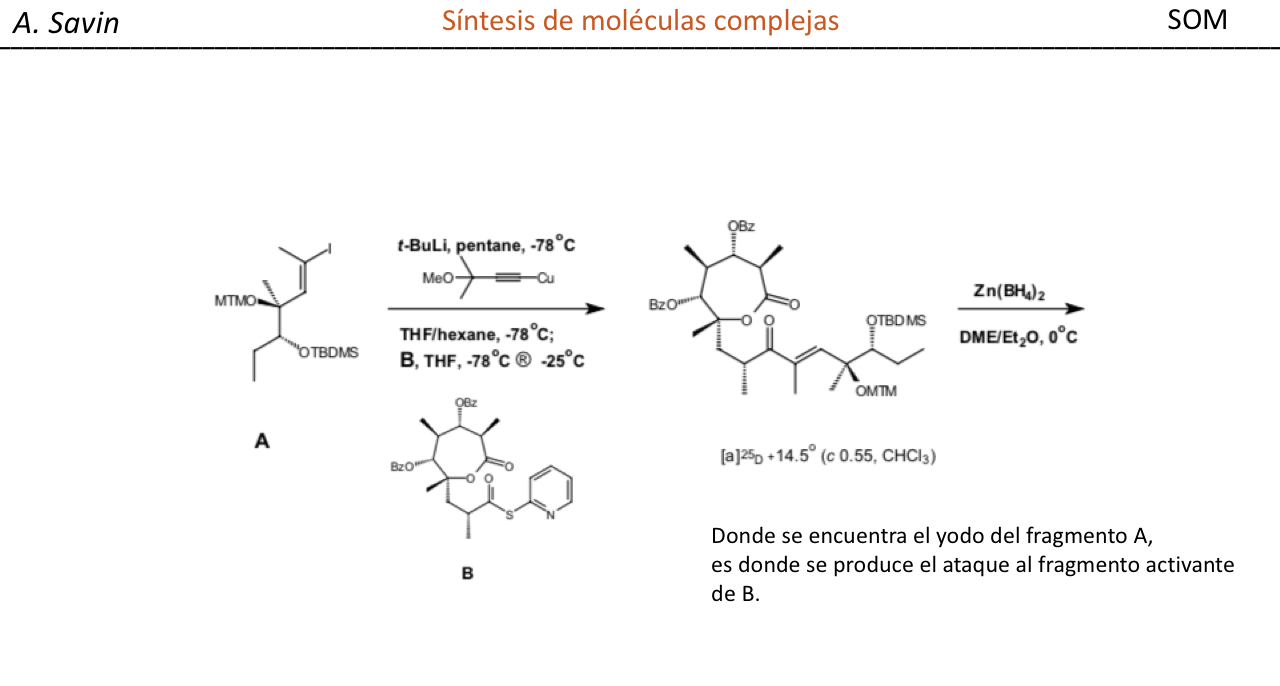
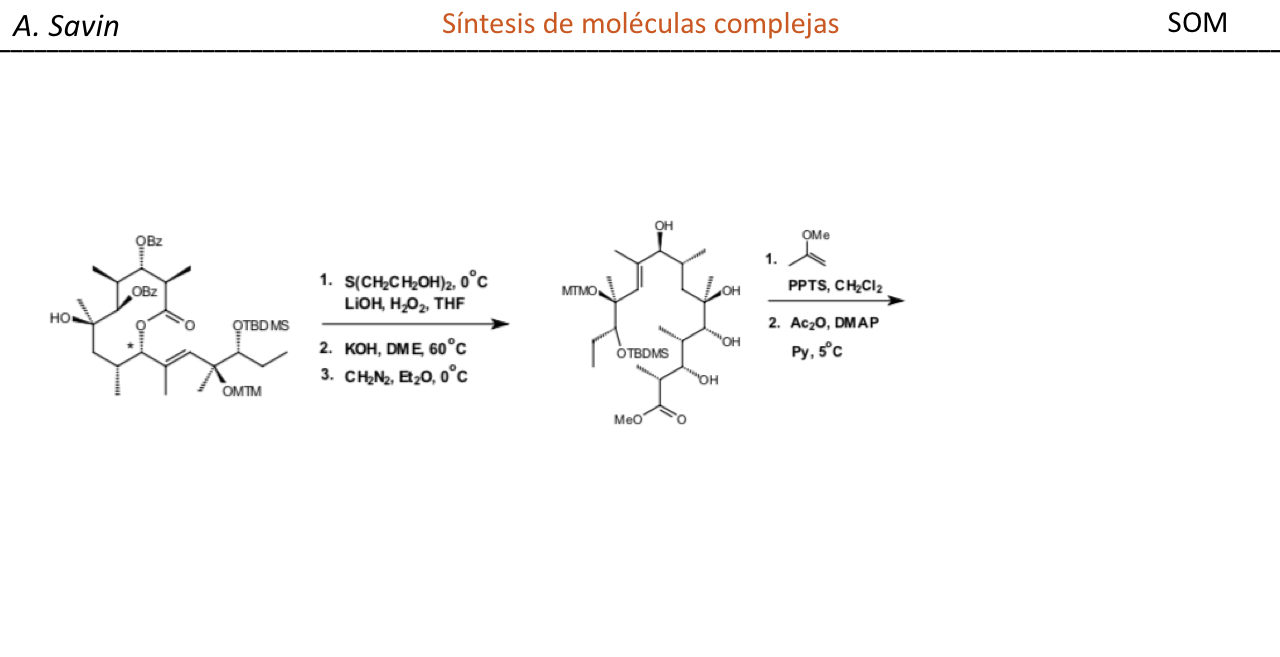


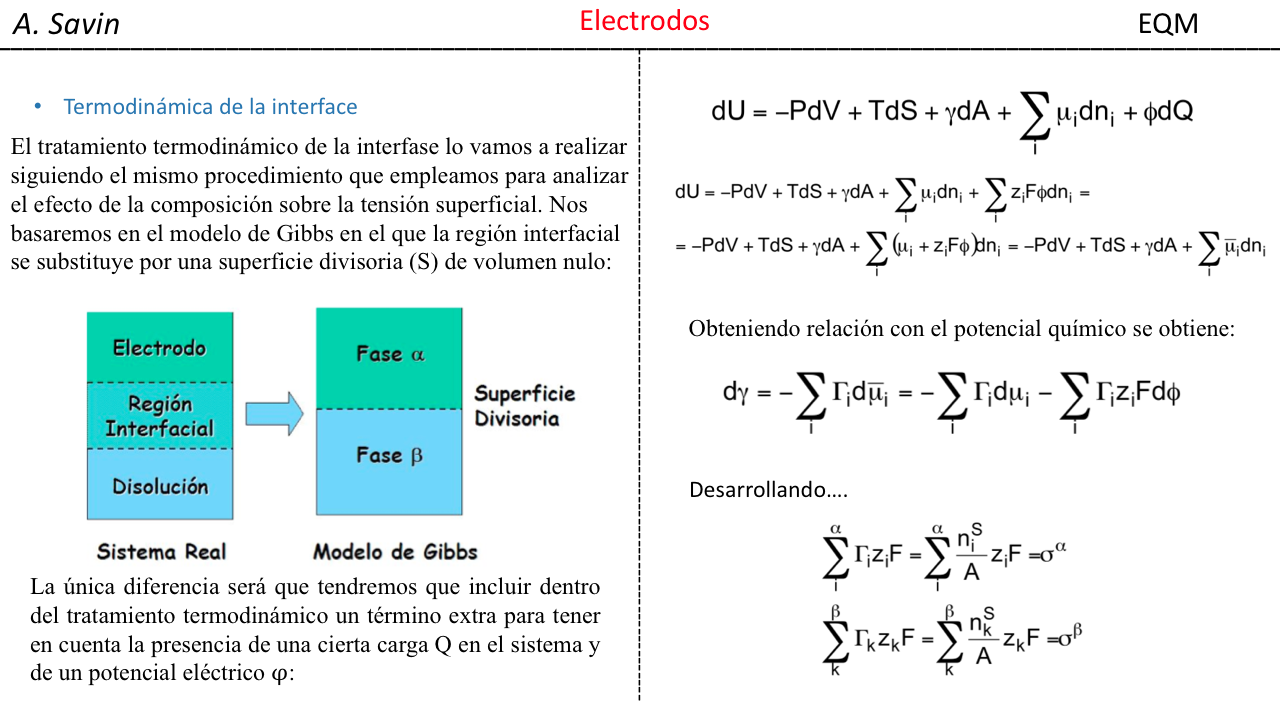
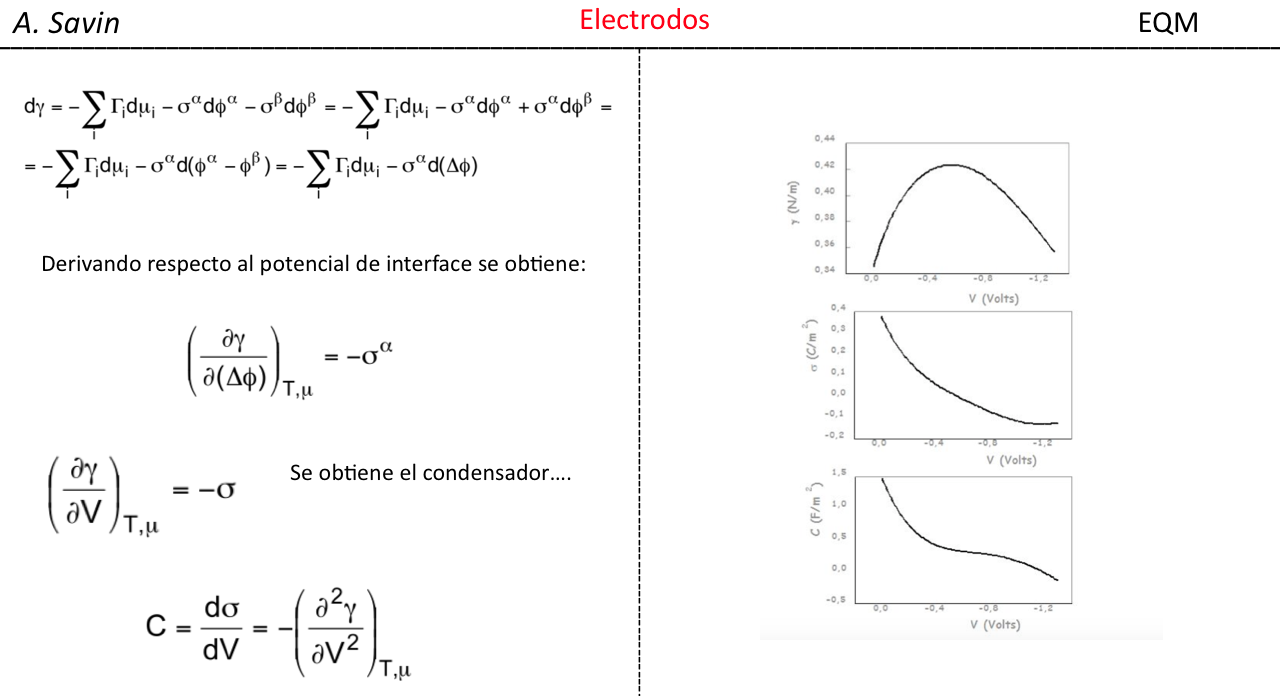

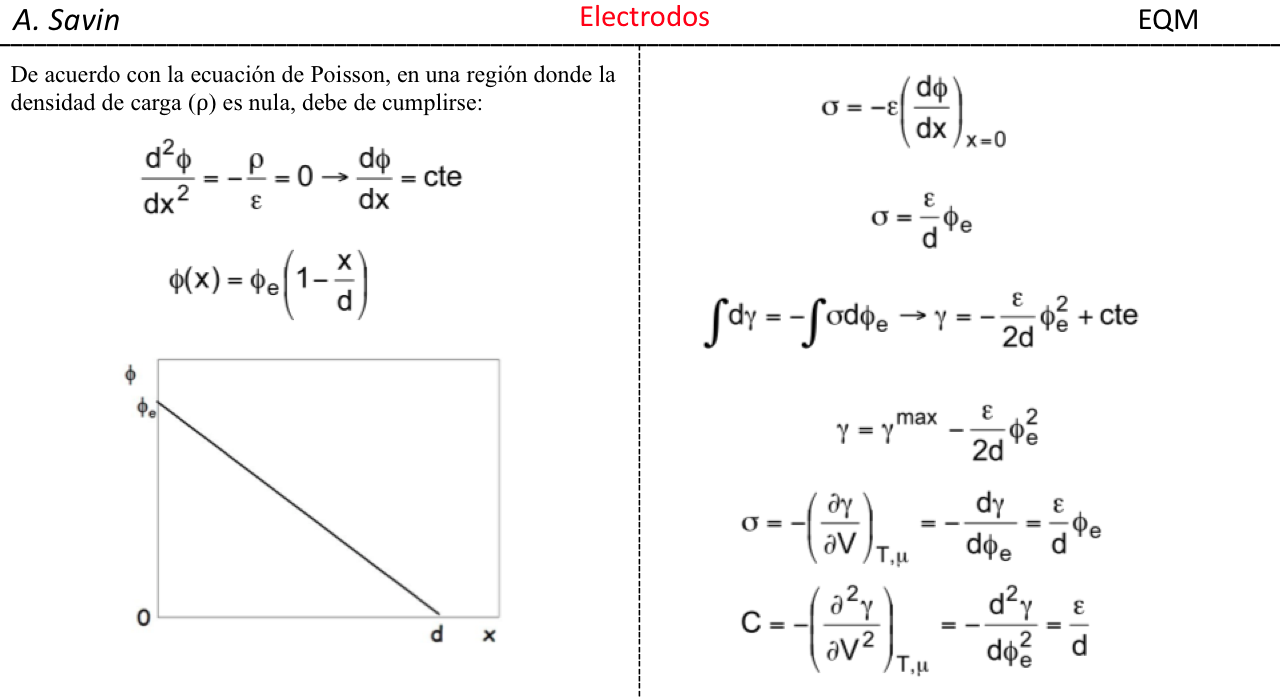
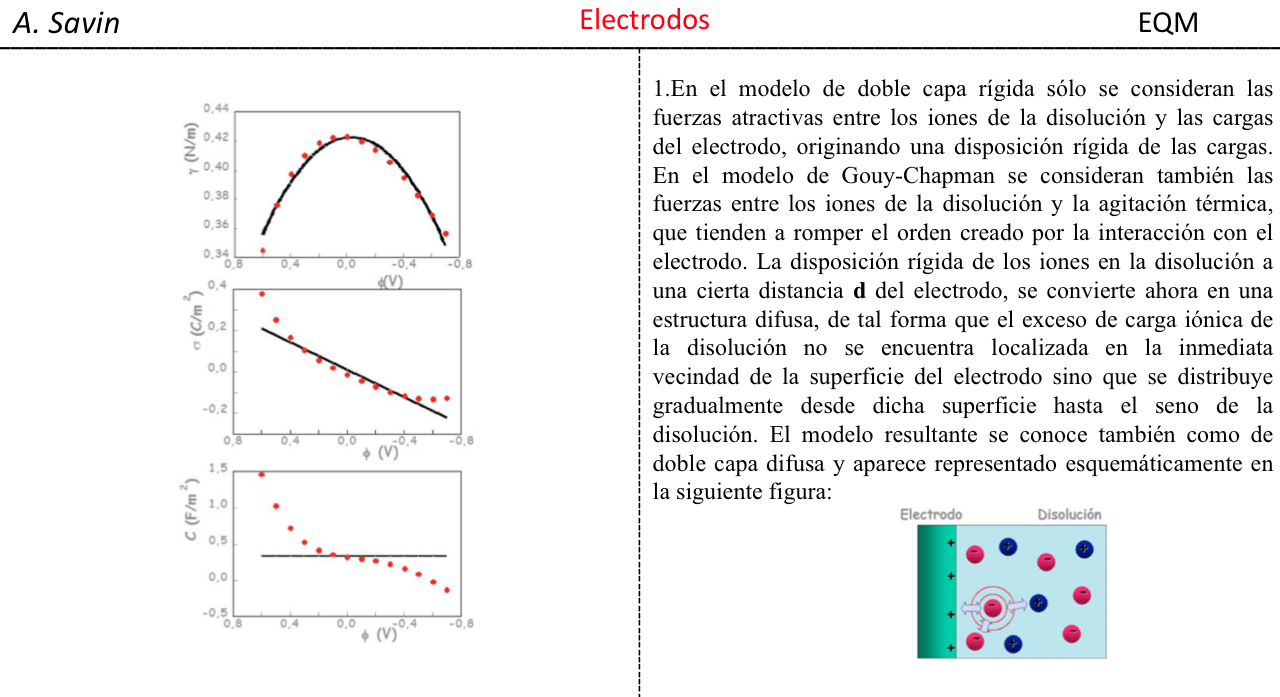




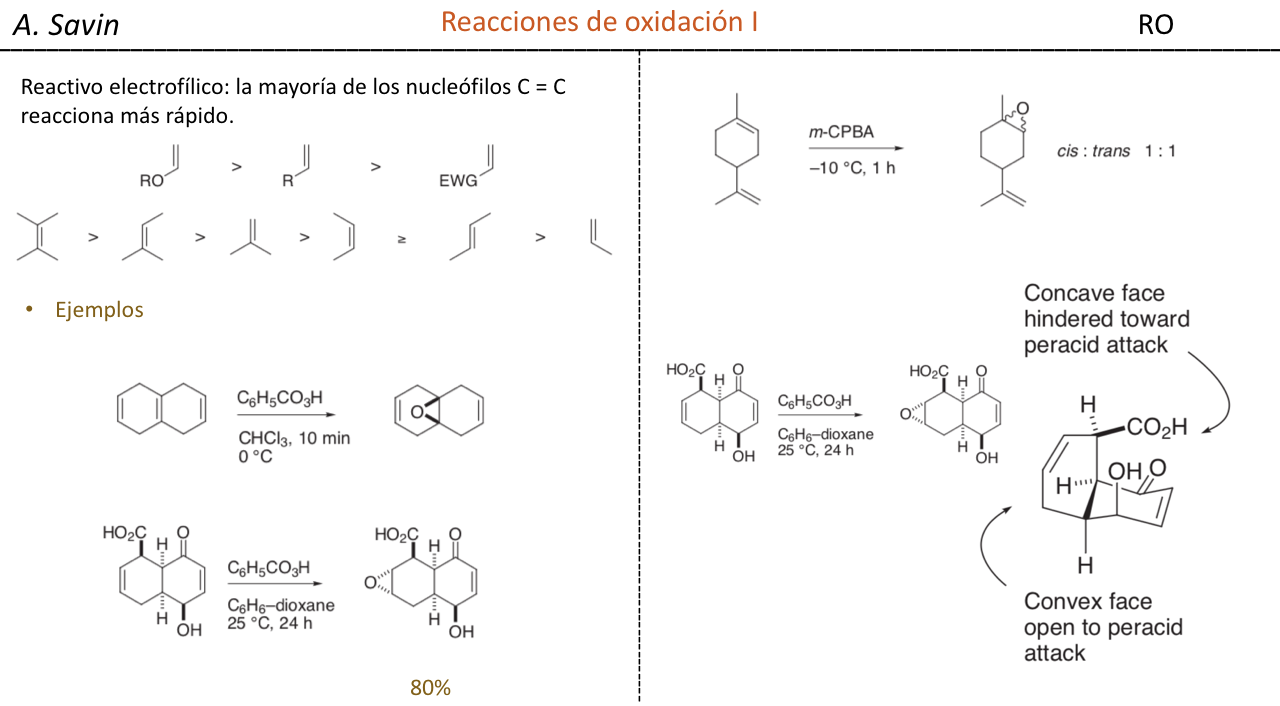
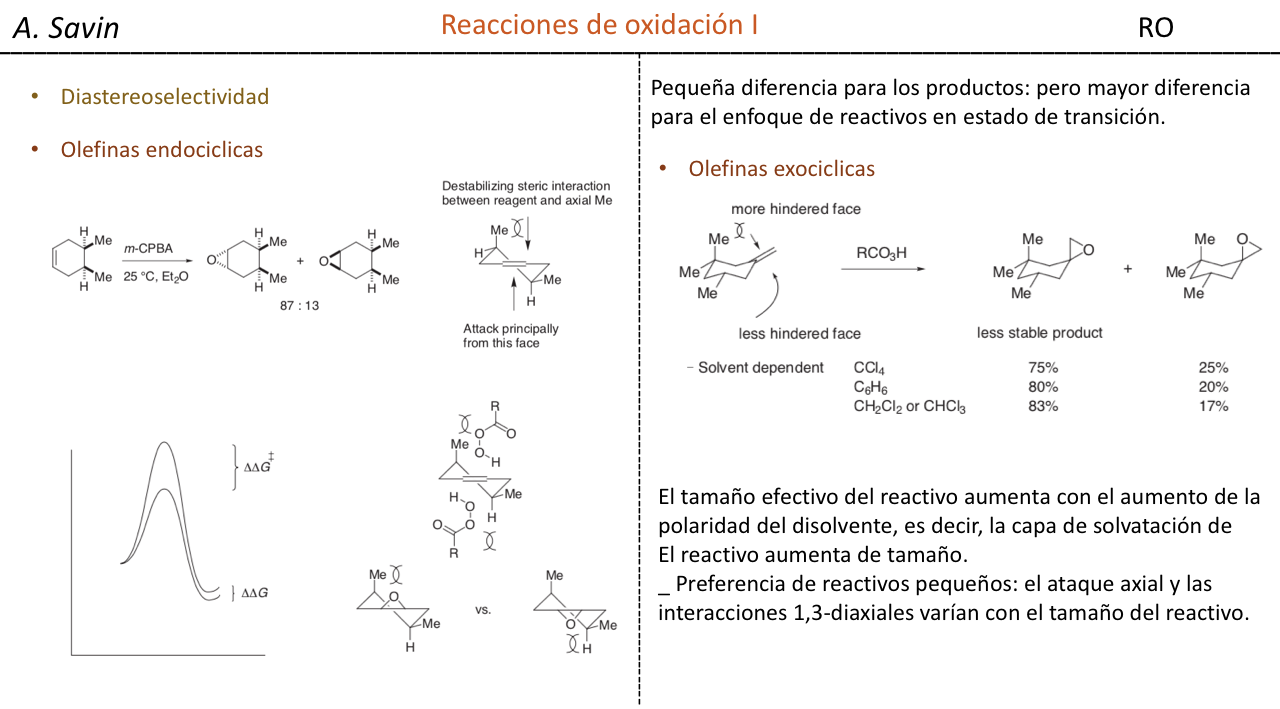
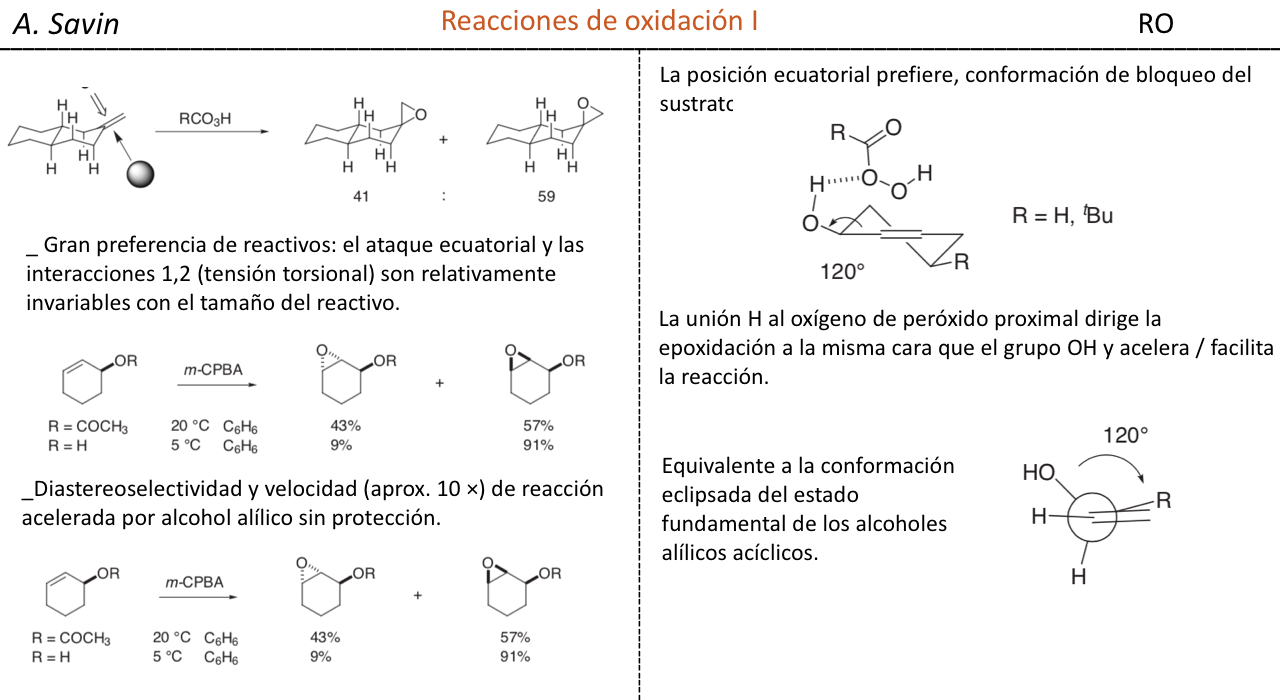
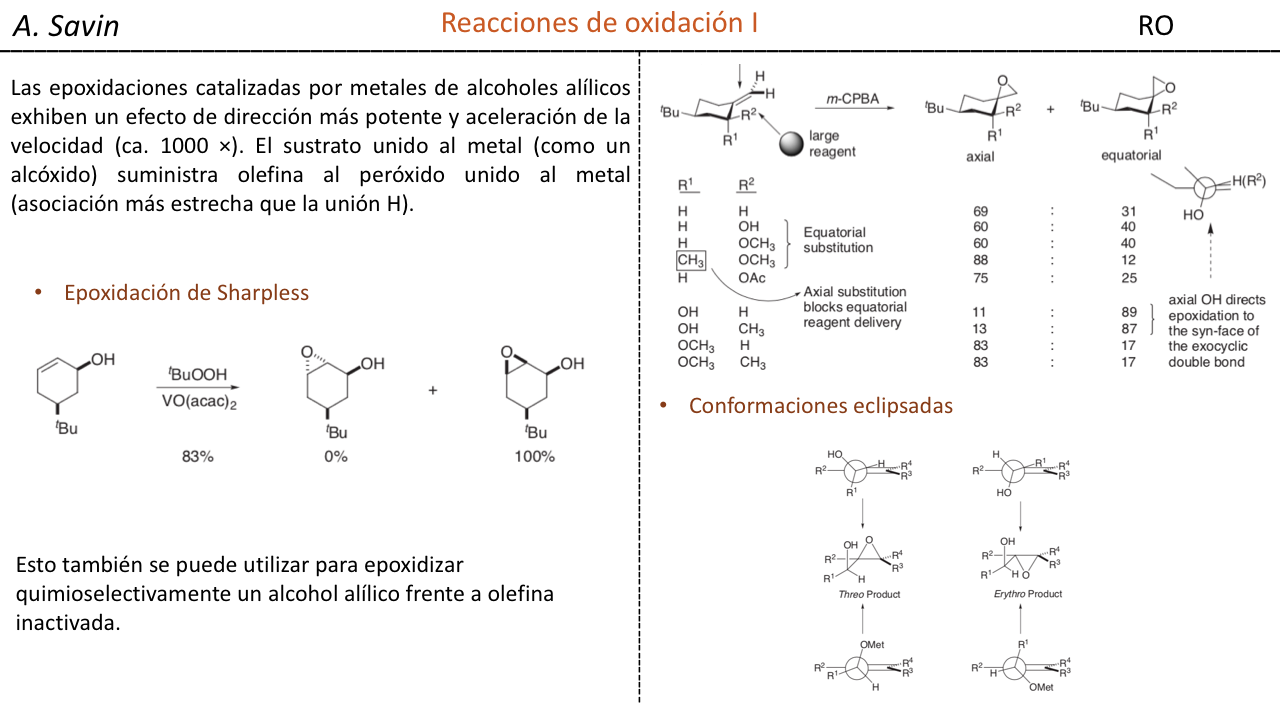
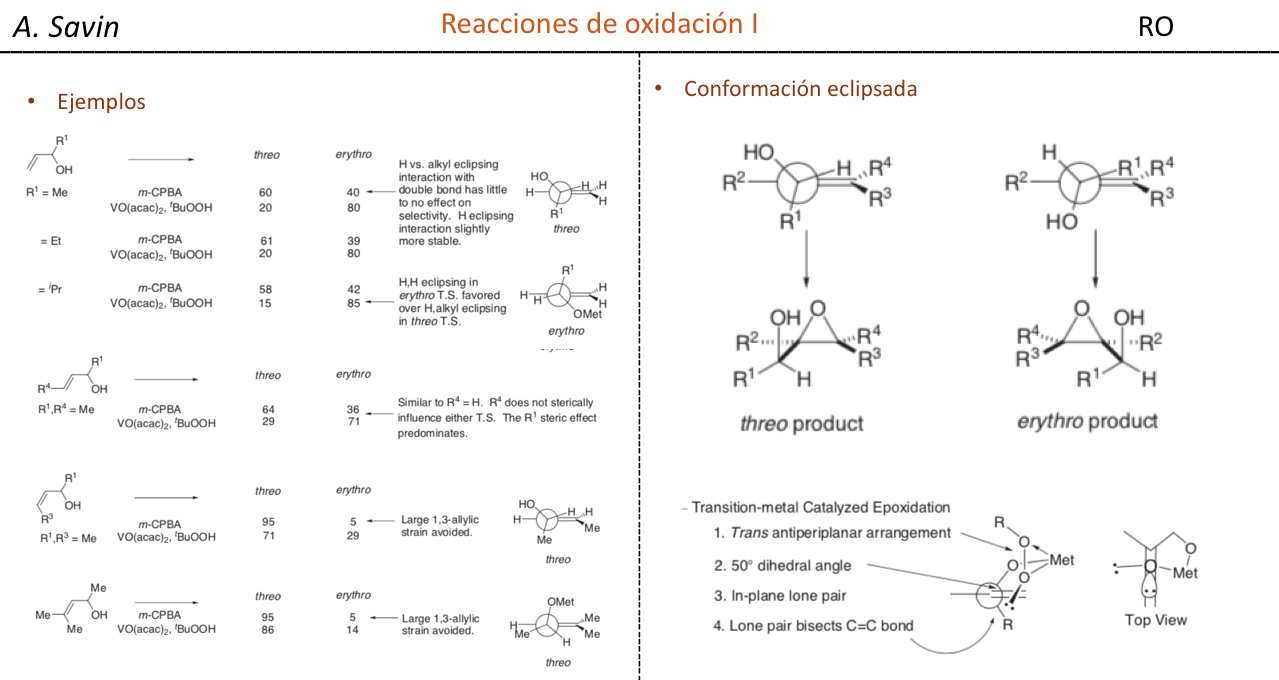
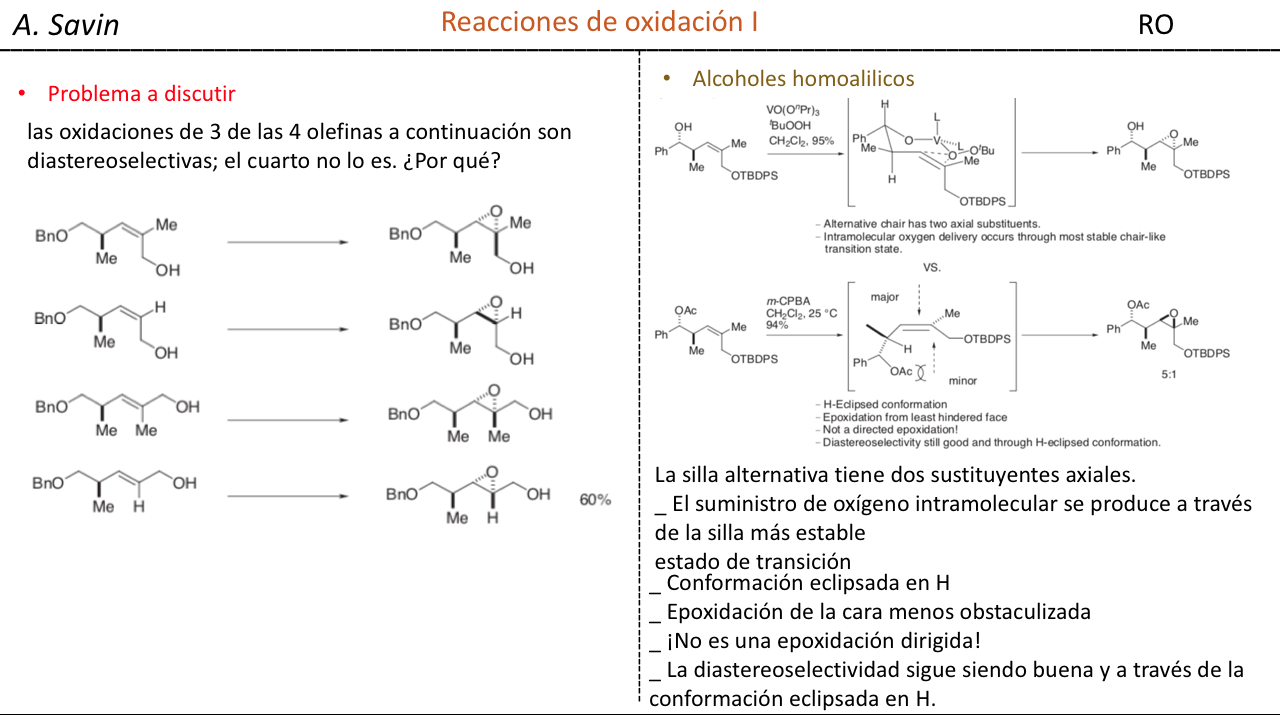
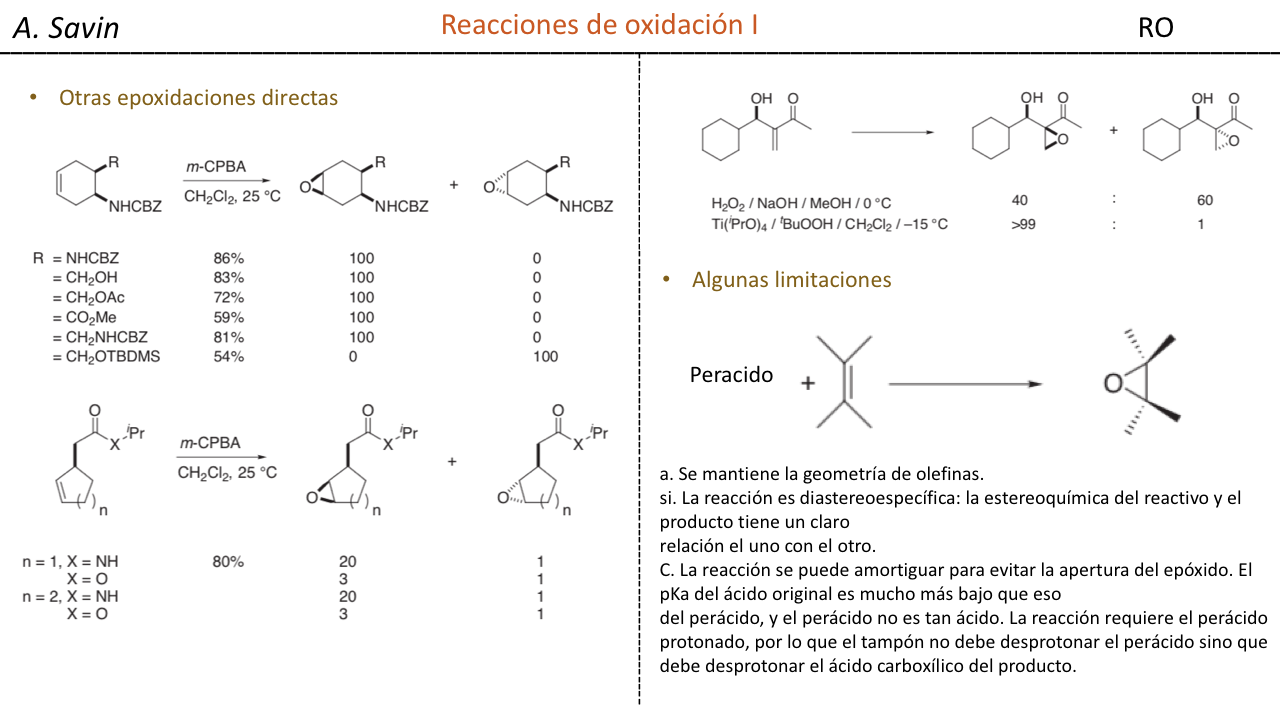
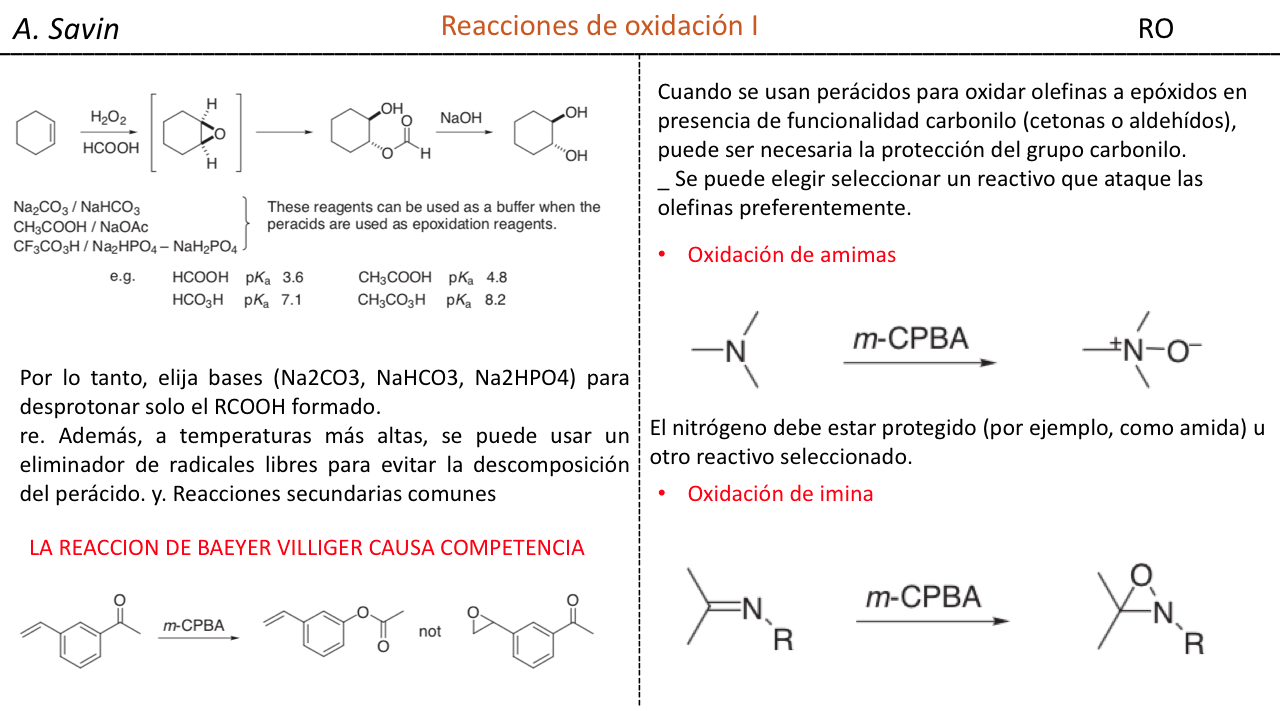
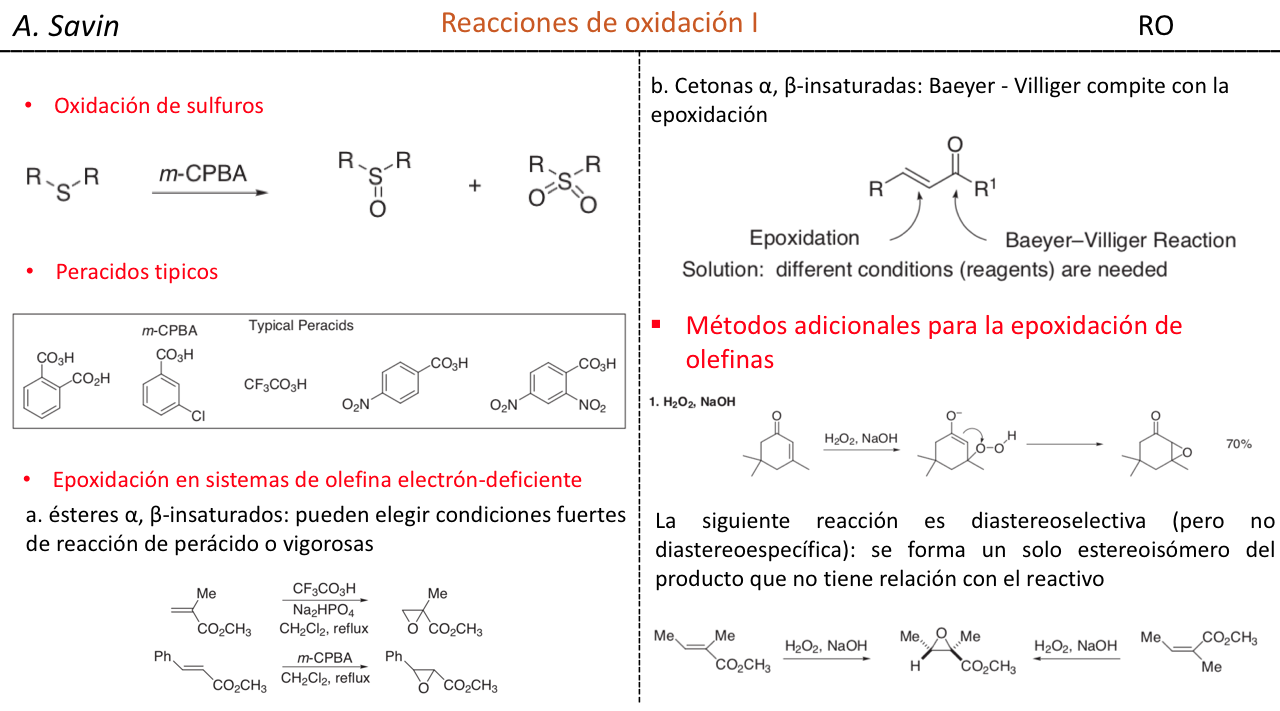
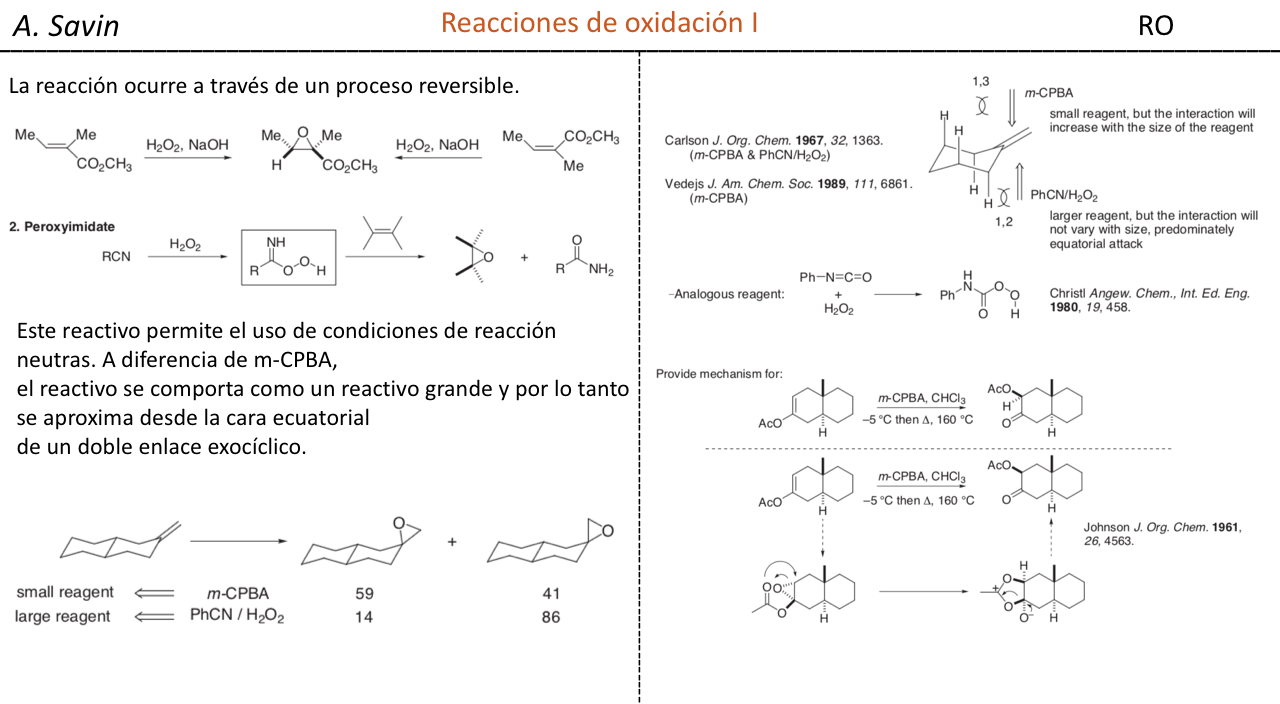
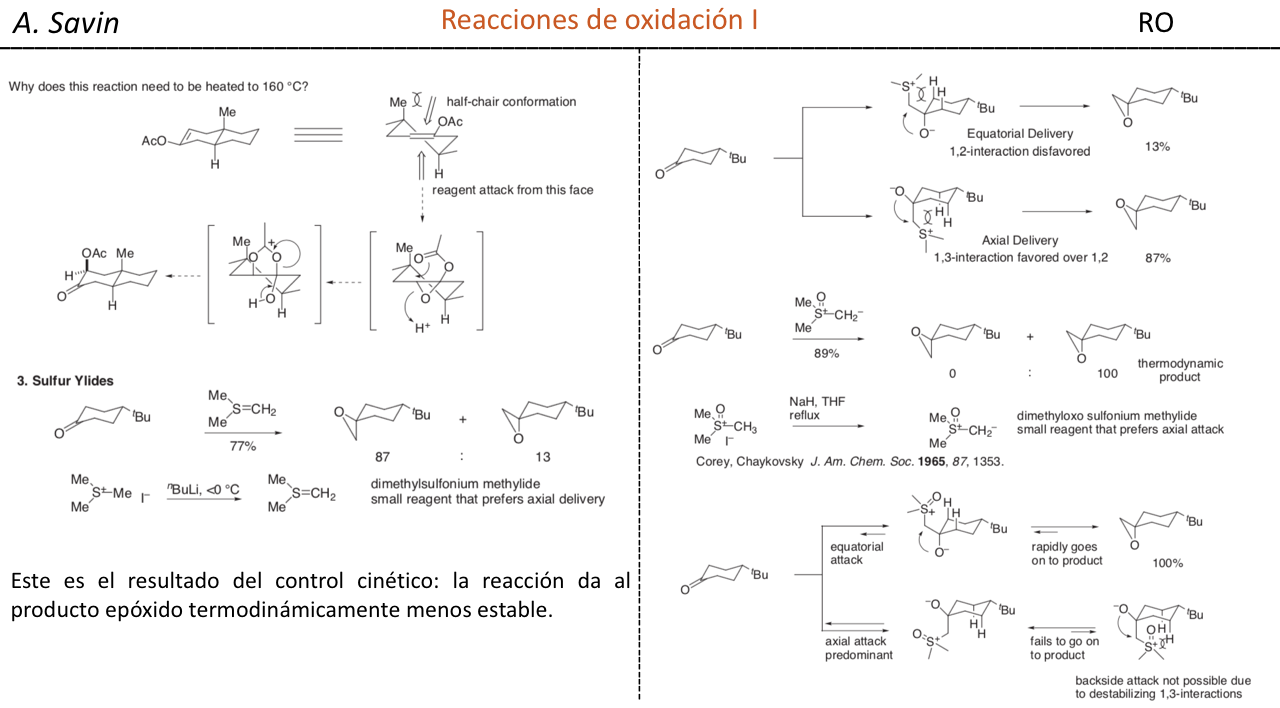
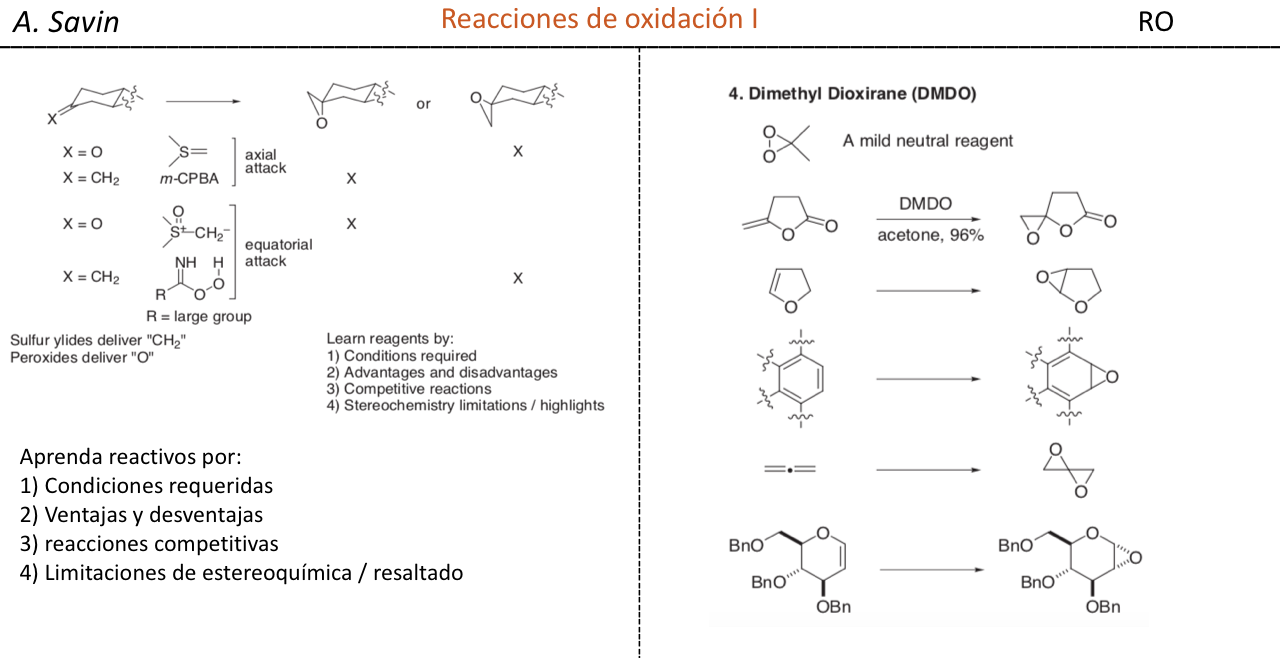

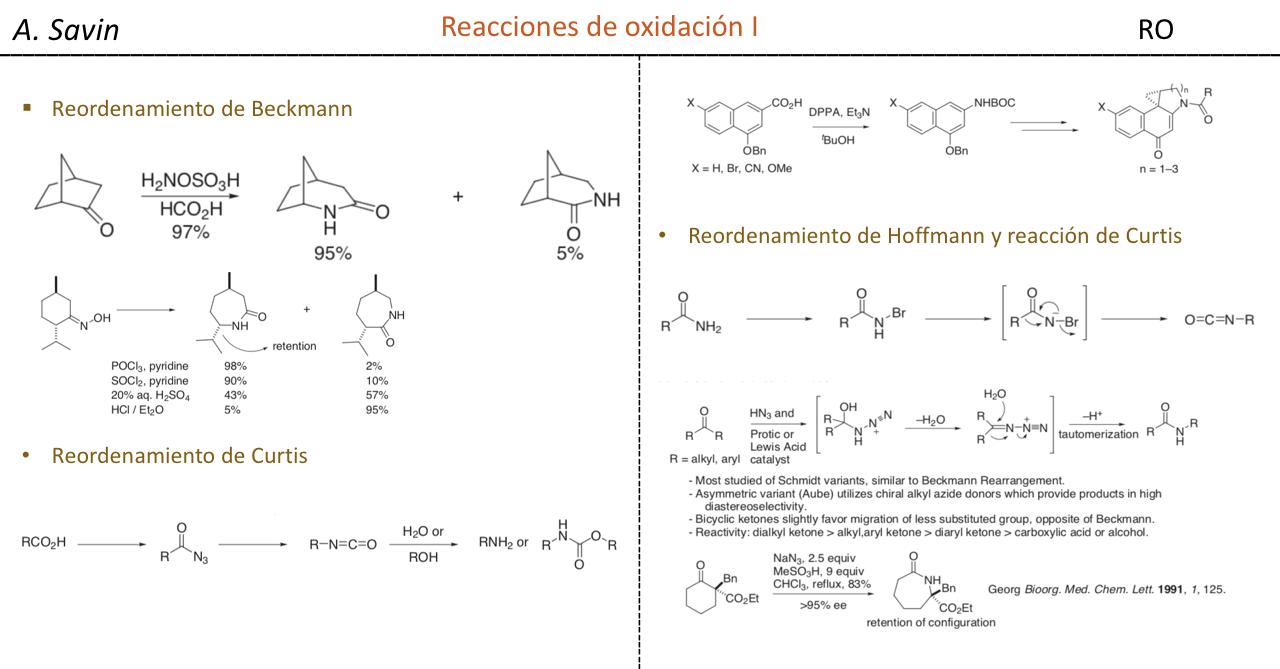
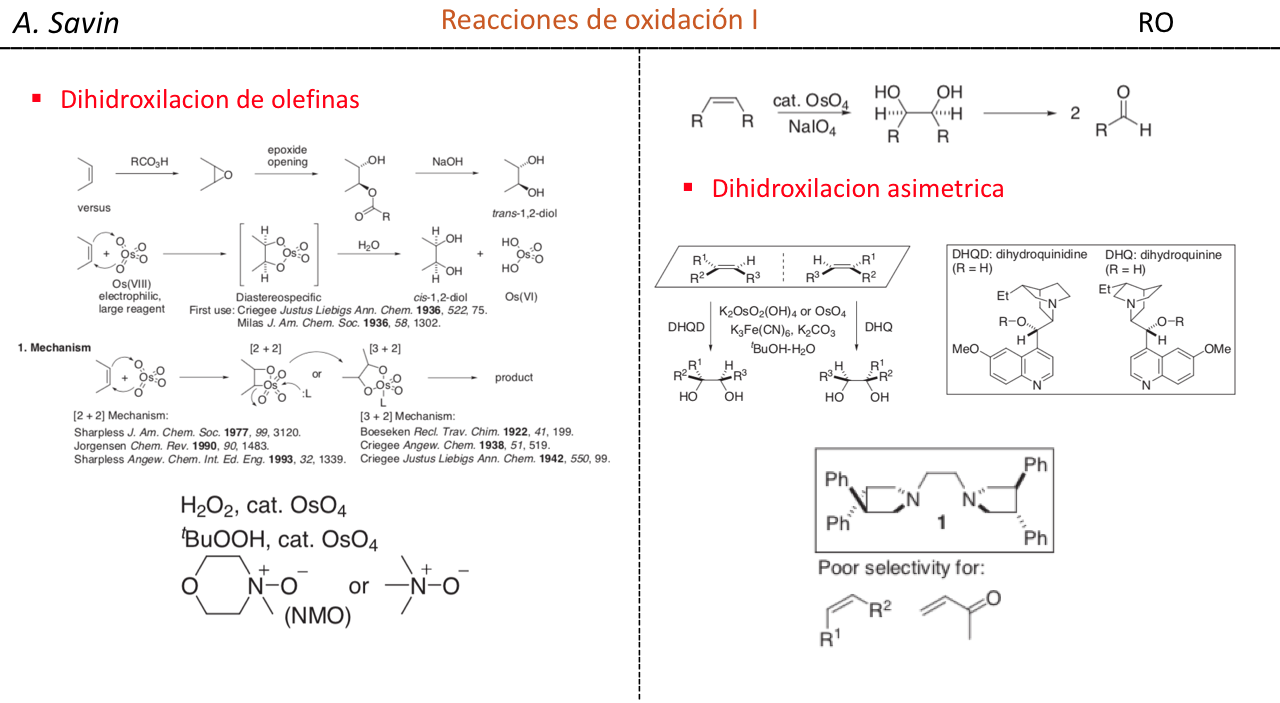
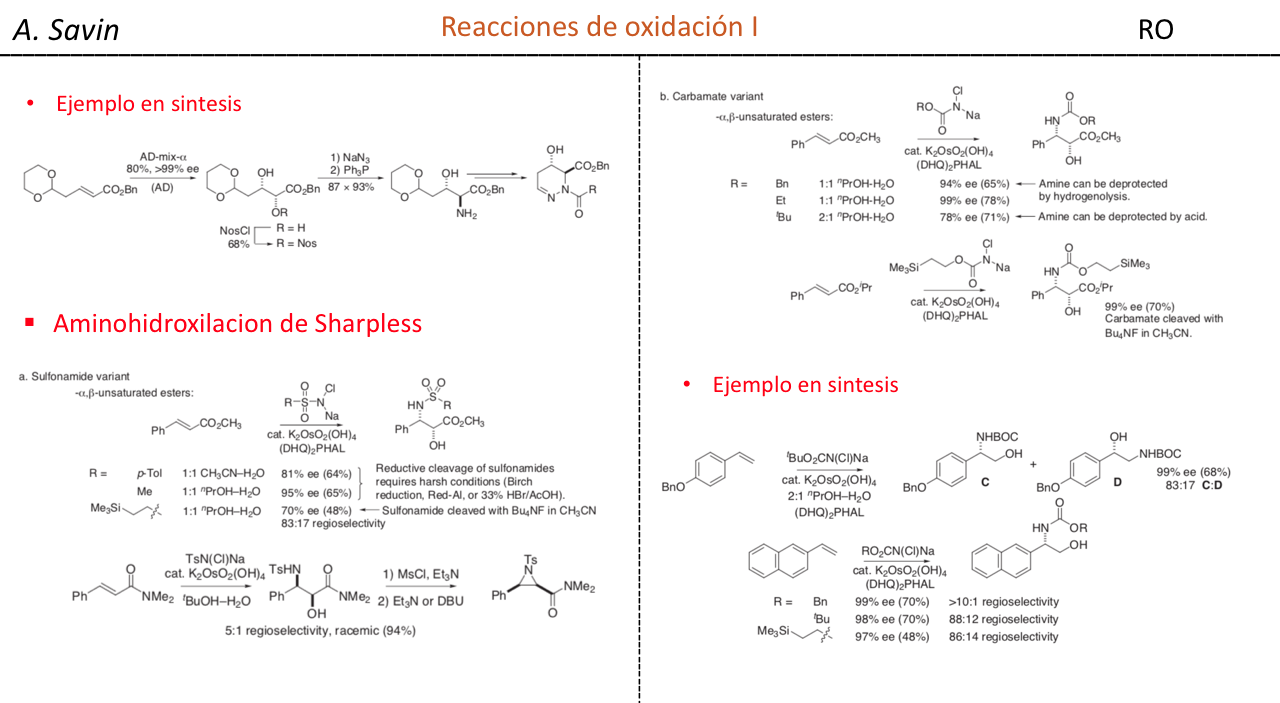
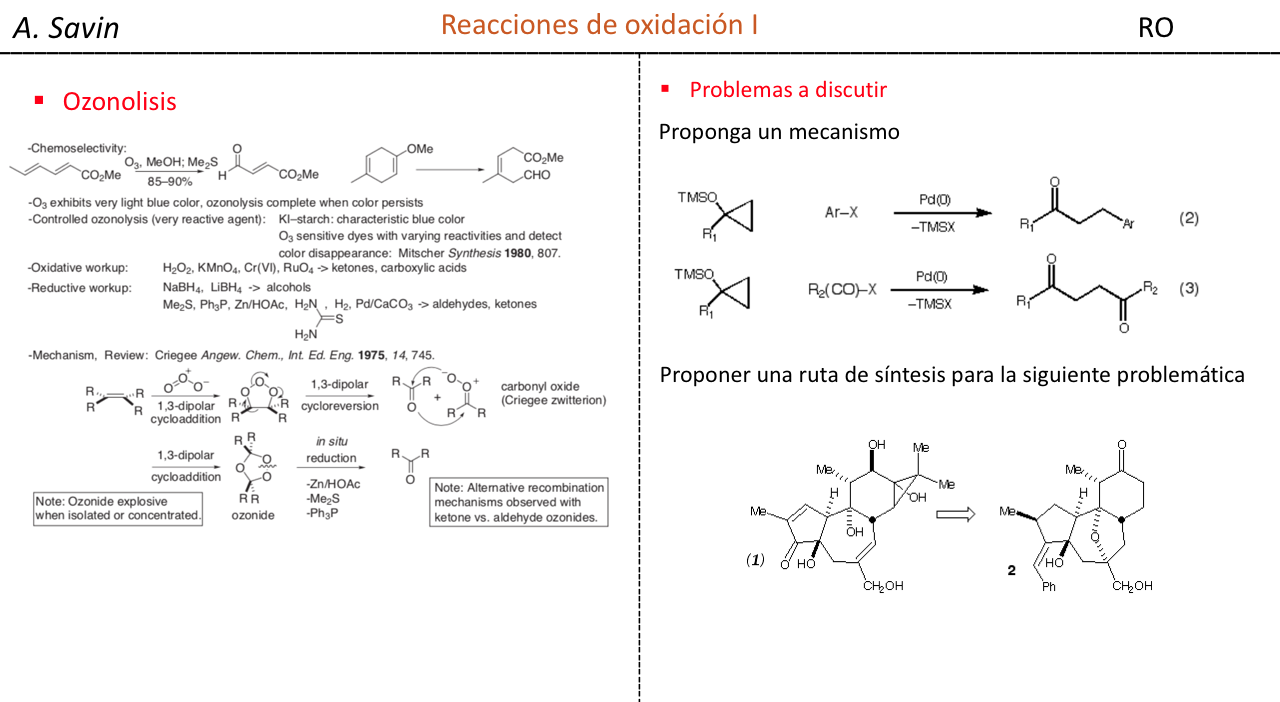


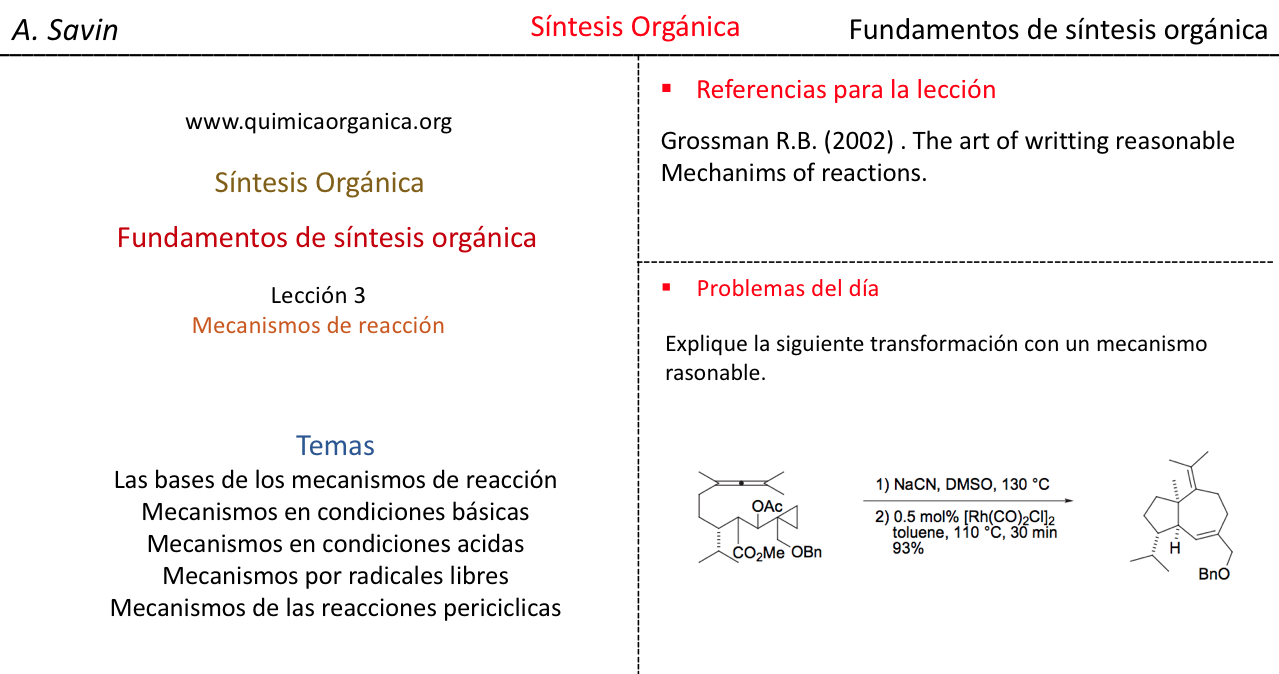
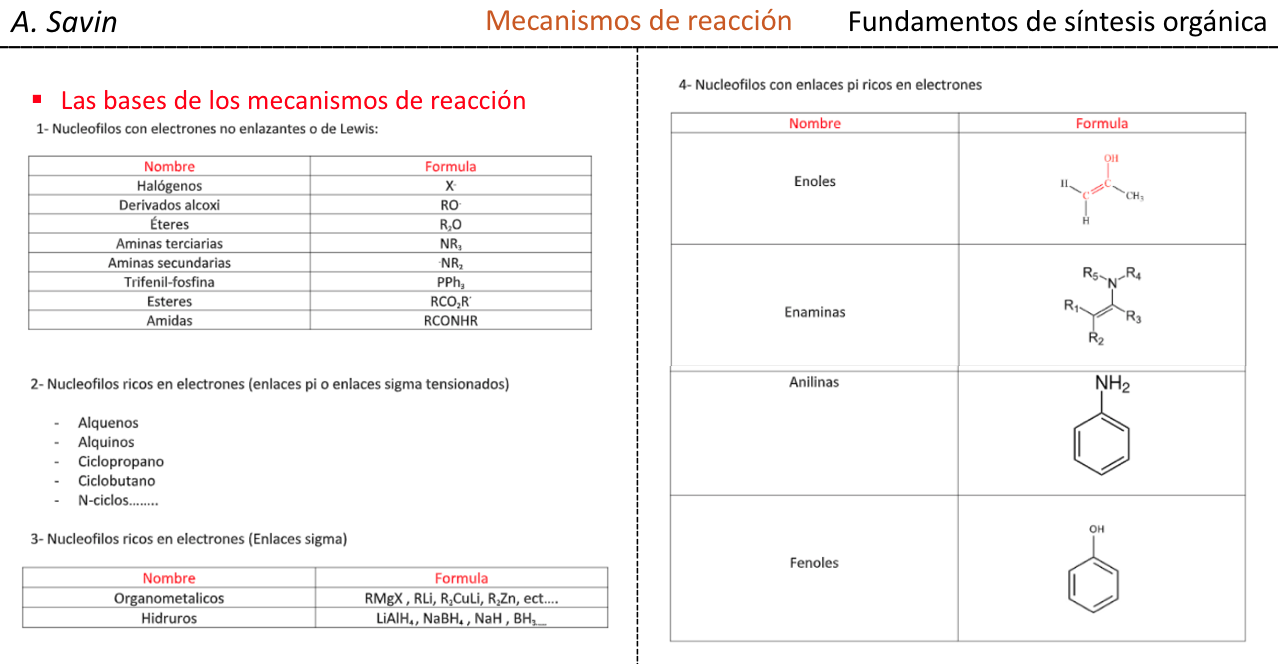
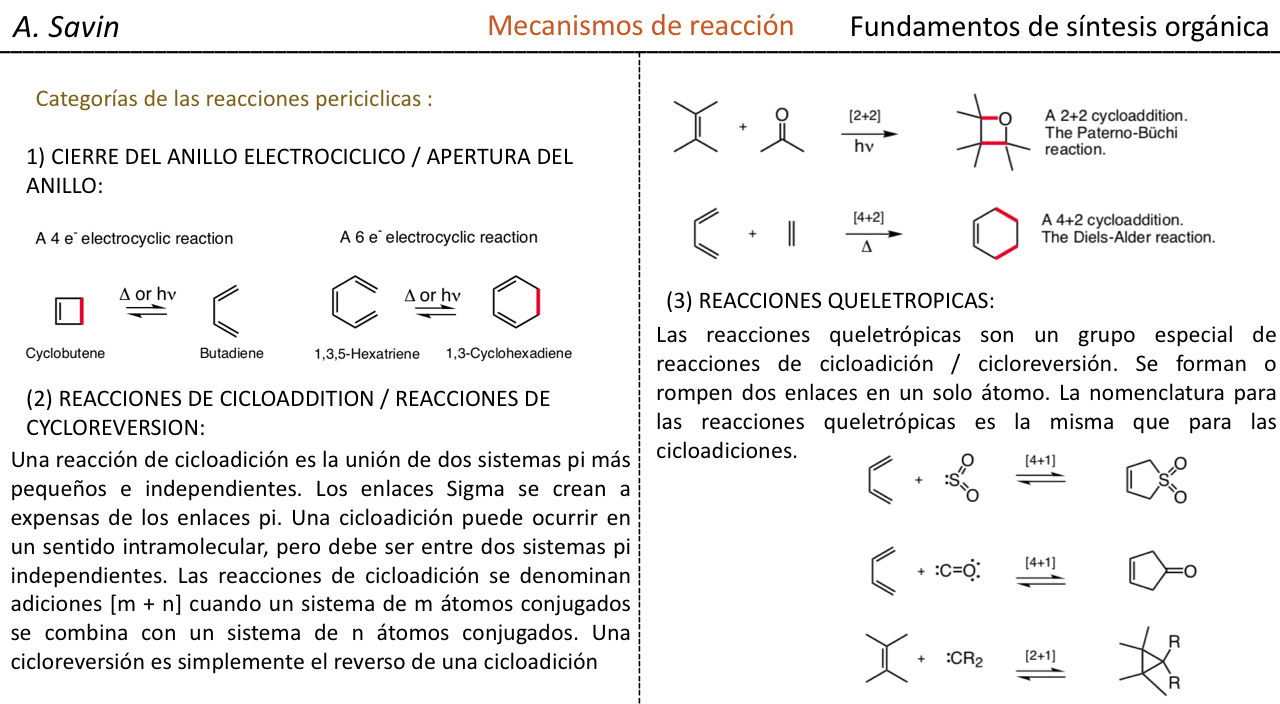
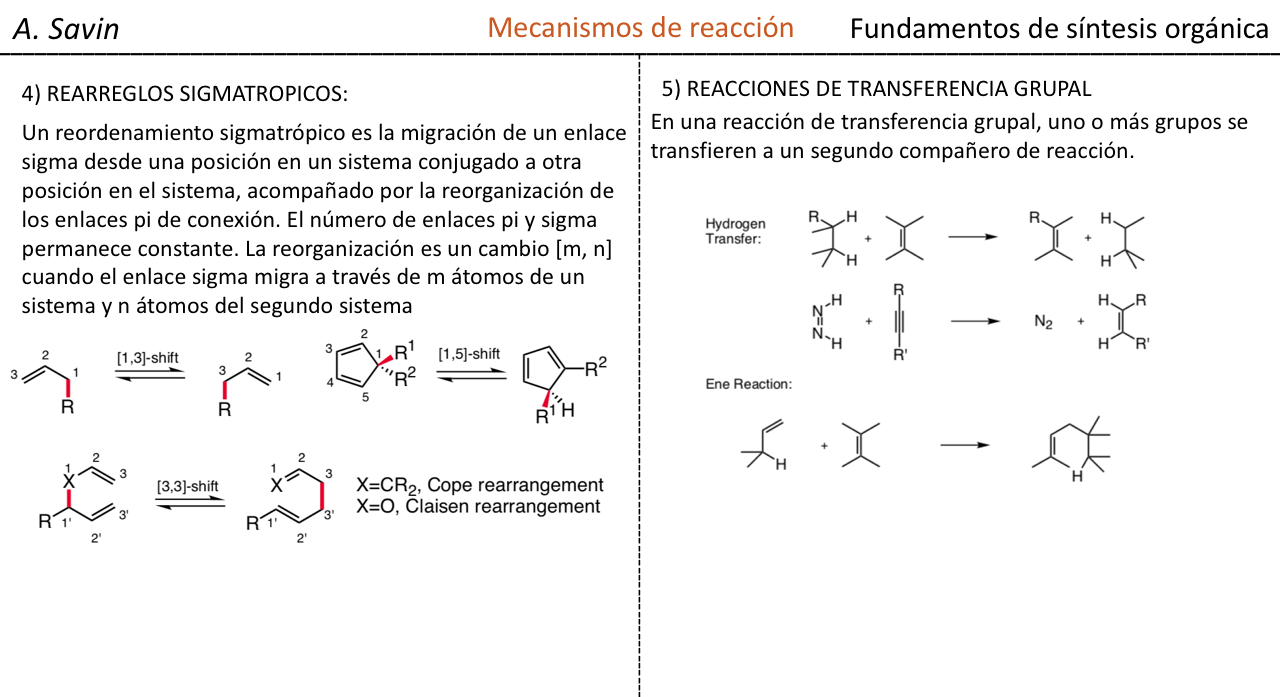
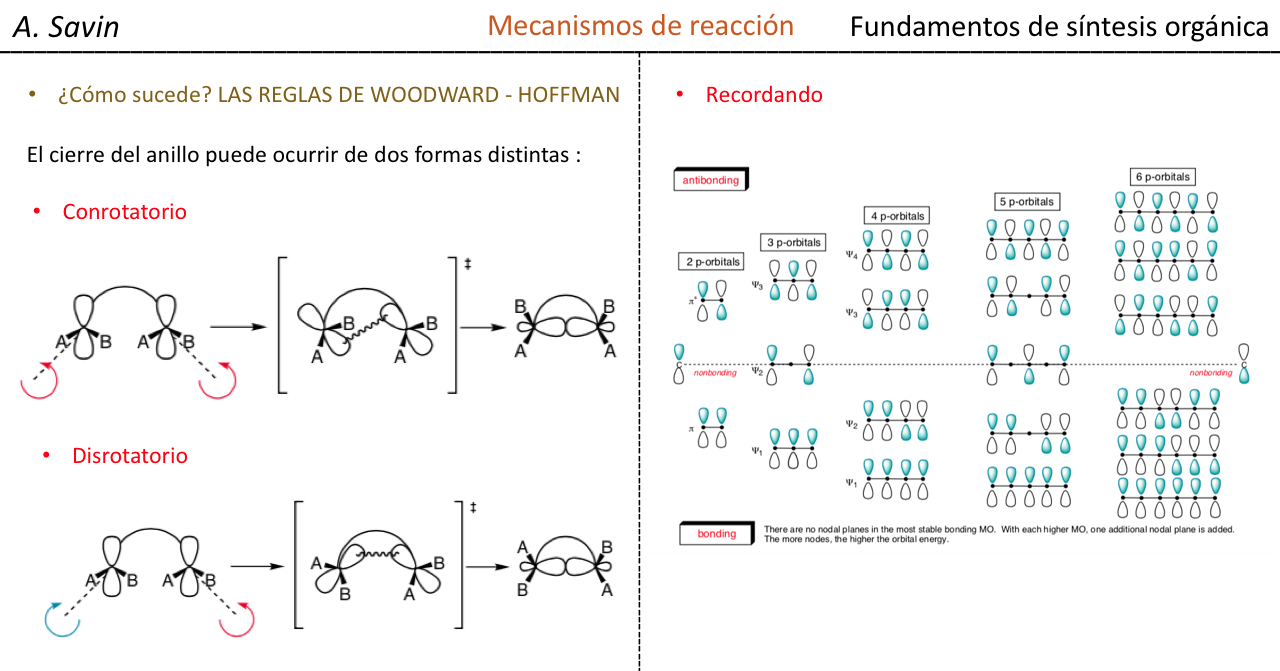
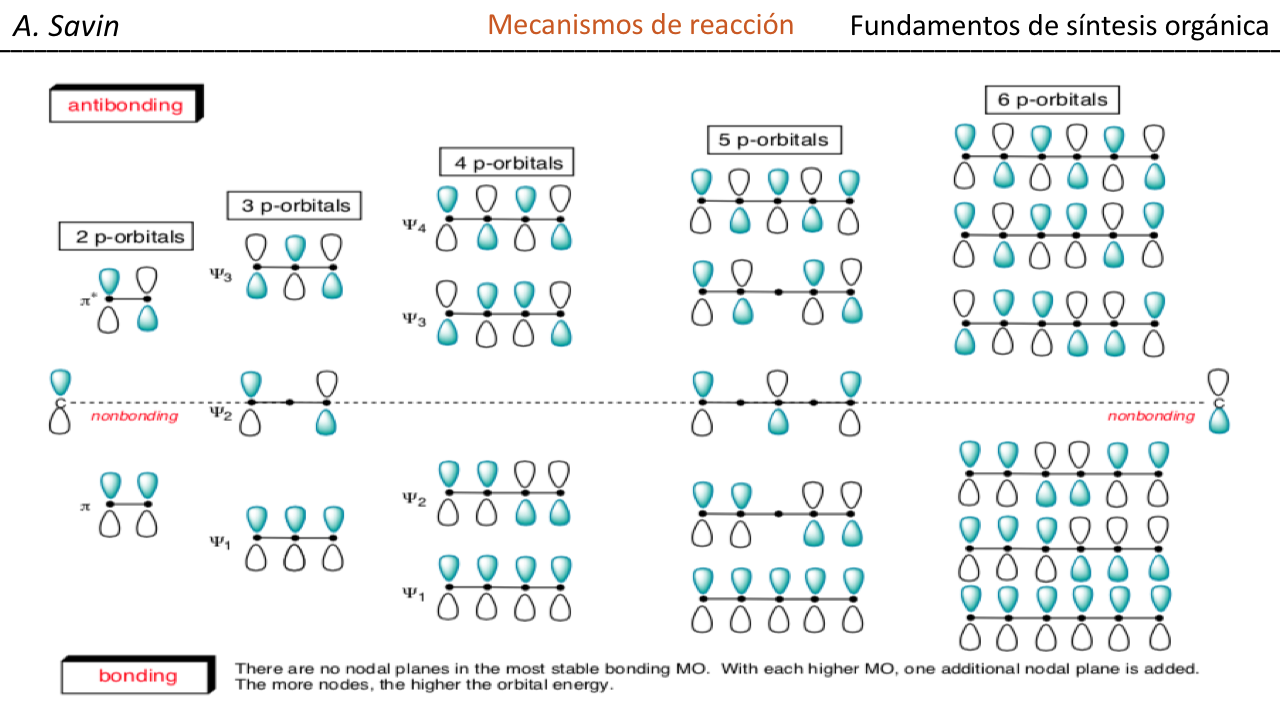
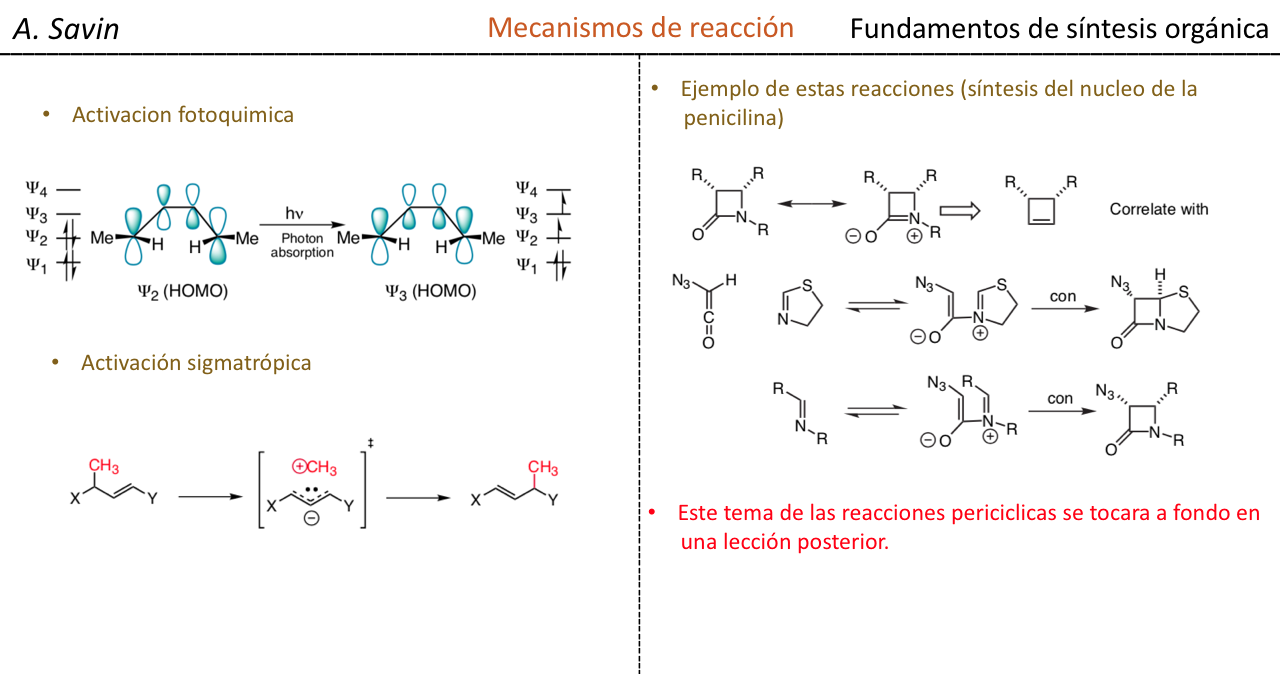
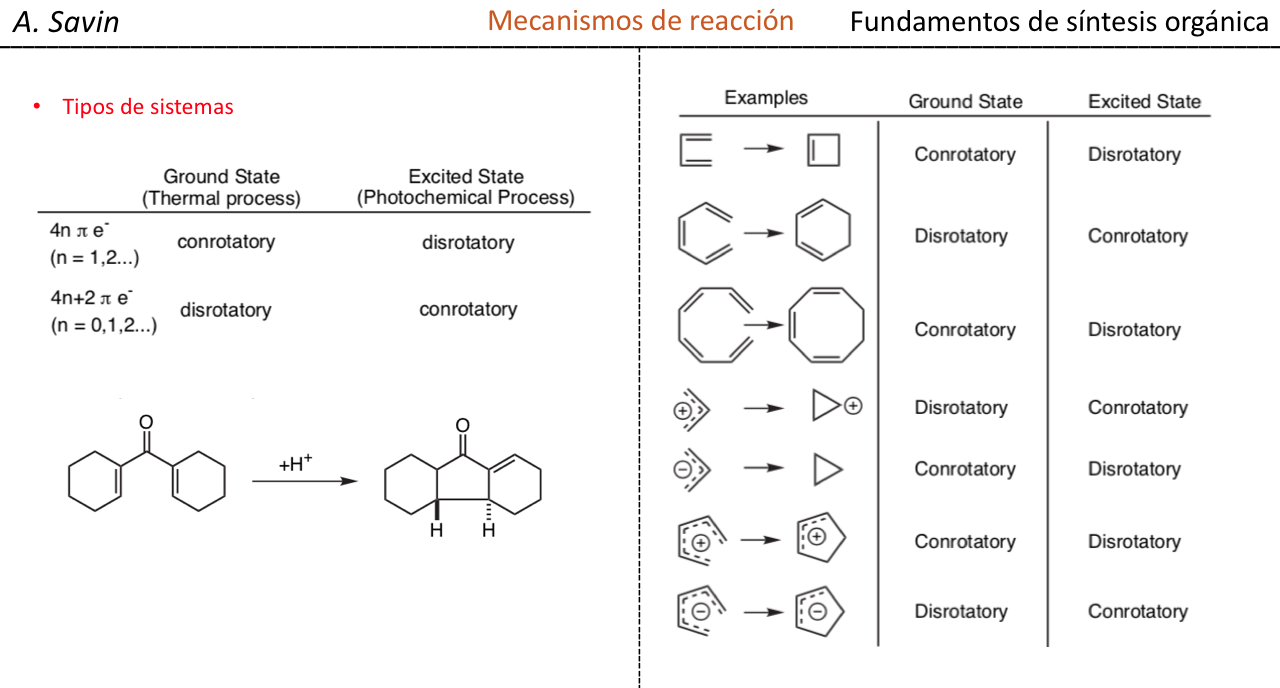



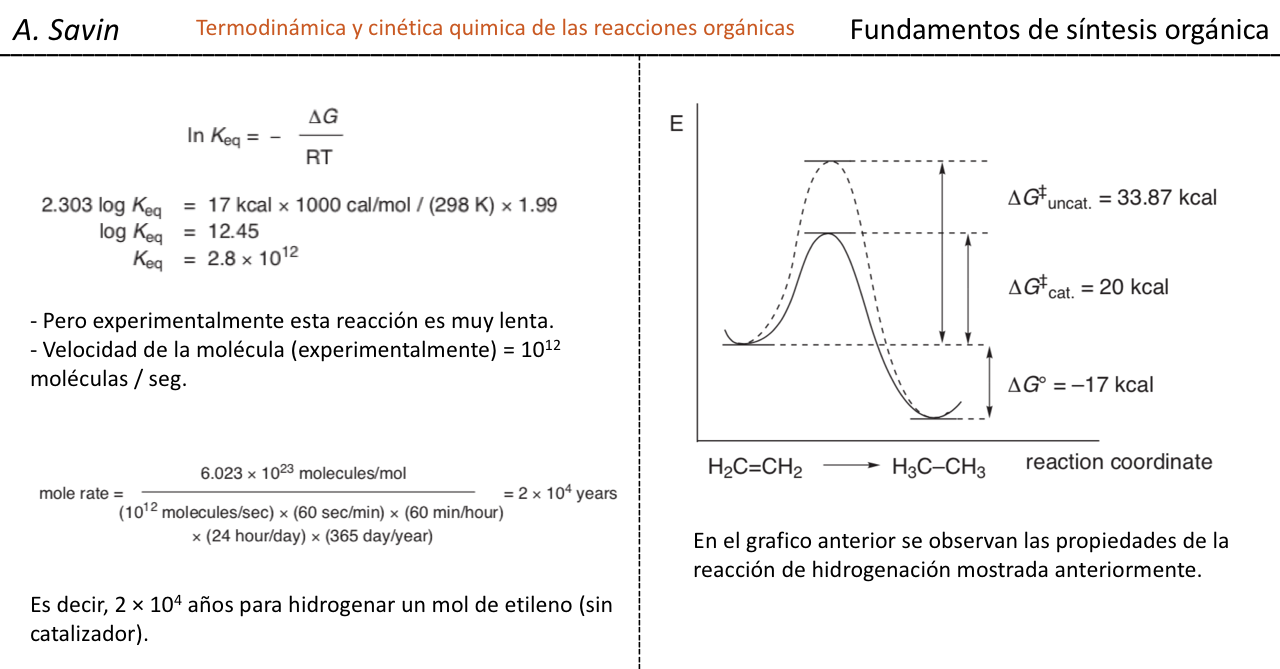

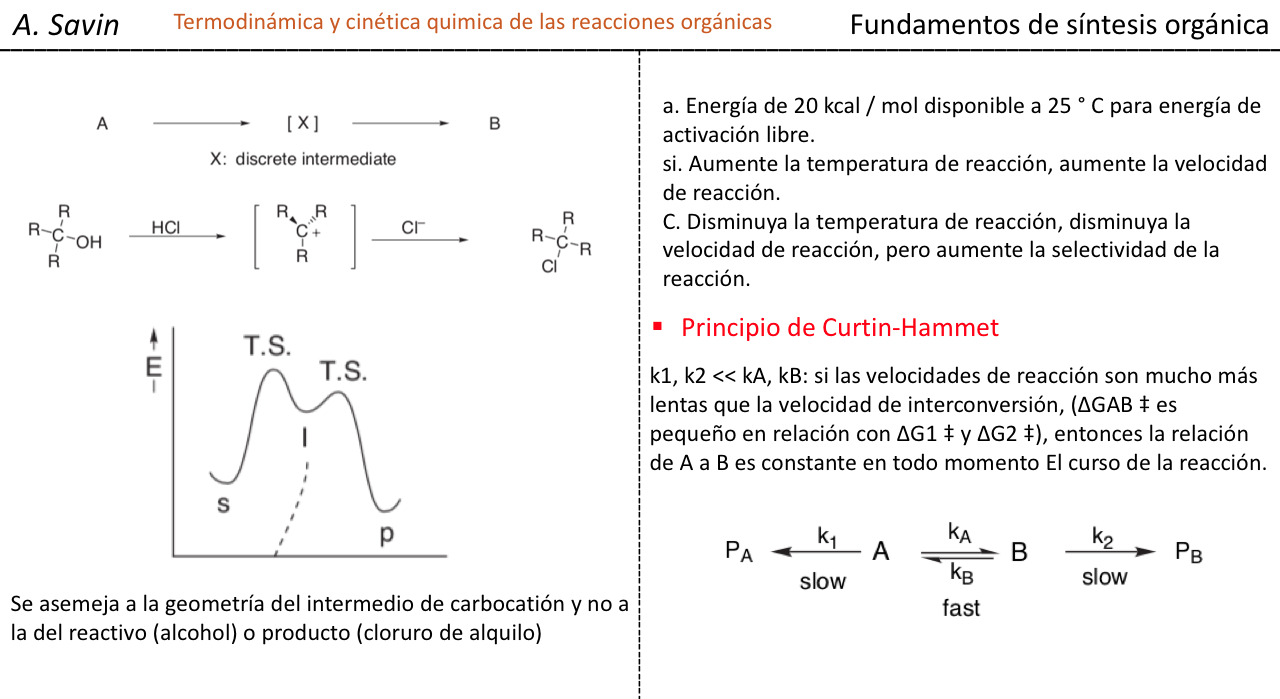
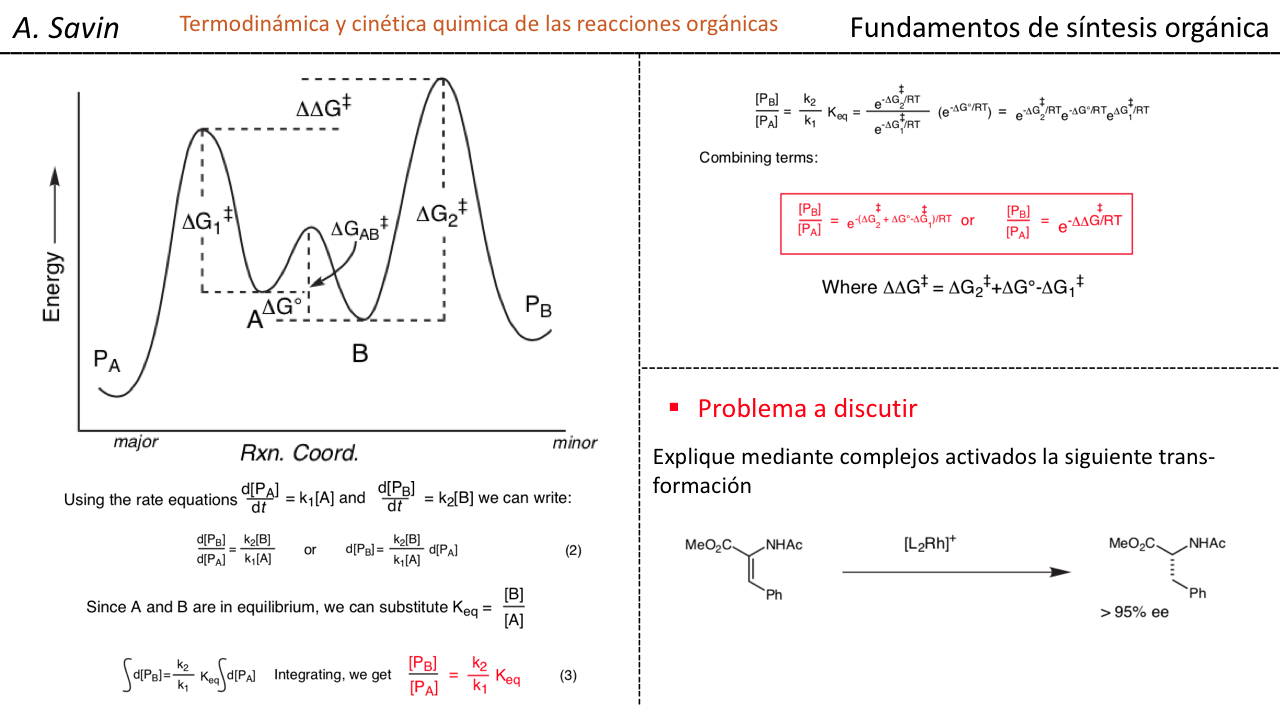
Efecto de la conjugación en la banda de Tensión C=O
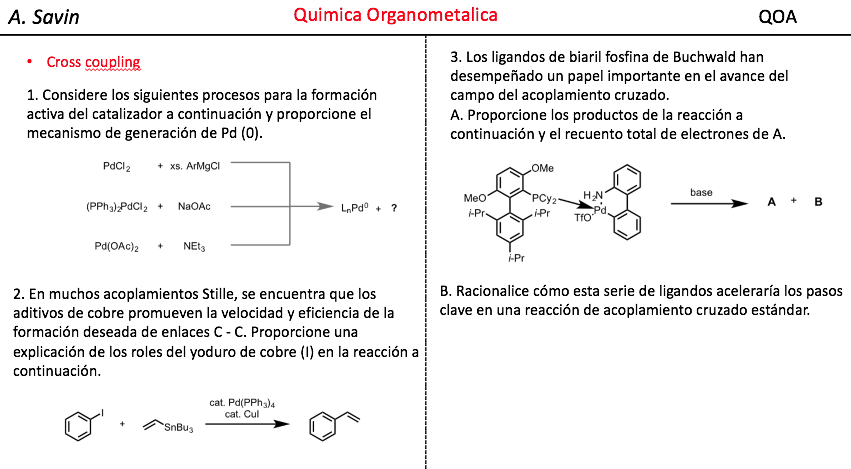
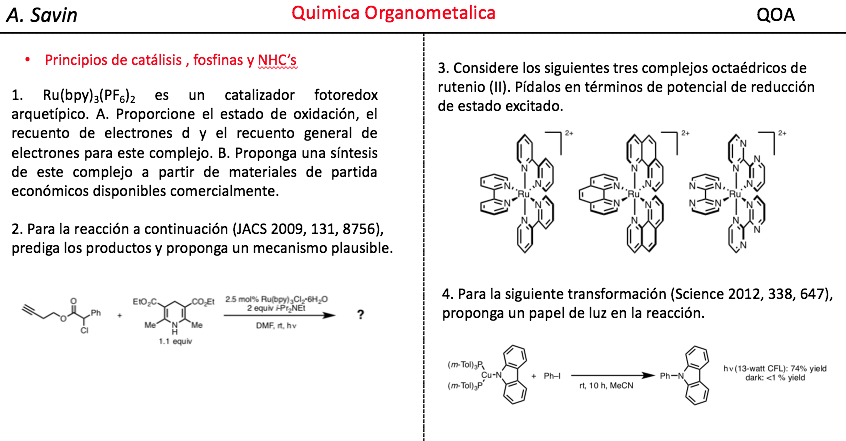
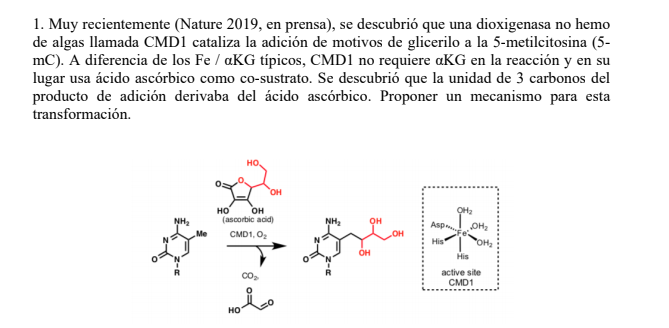
![]() ¿Te cuesta entender la Química Orgánica?
¿Te cuesta entender la Química Orgánica?
Cursos de Química Orgánica para los Grados en Química, Ingeniería Química, Biotecnología y Farmacia de las Universidades españolas.
Material específico para cada Universidad con teoría, ejercicios y exámenes resueltos en vídeo, creado por Germán Fernández. Soporte para dudas por WhatsApp. .
Más información en www.foroquimico.com
Si prefieres un curso de química orgánica tanto básica como avanzada te invito a acceder al canal: https://www.youtube.com/channel/UC_RiUaA2326jO9XozAA4q2g , en el que encontrarás más de 750 vídeos de teoría y ejercicios.
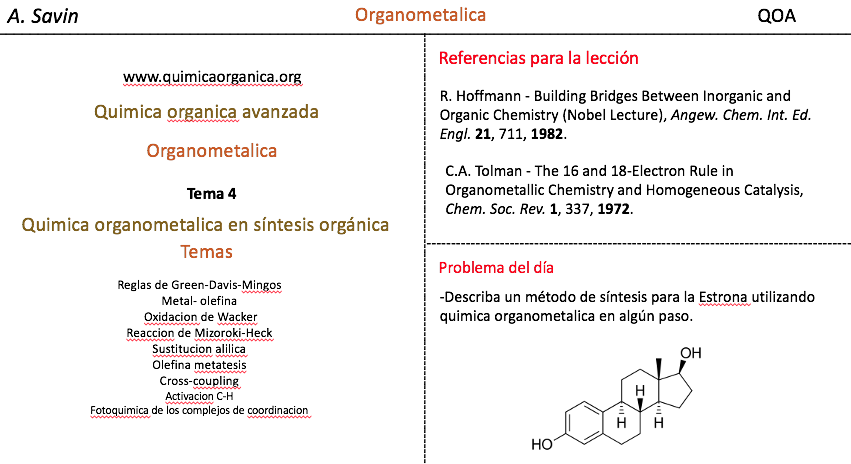
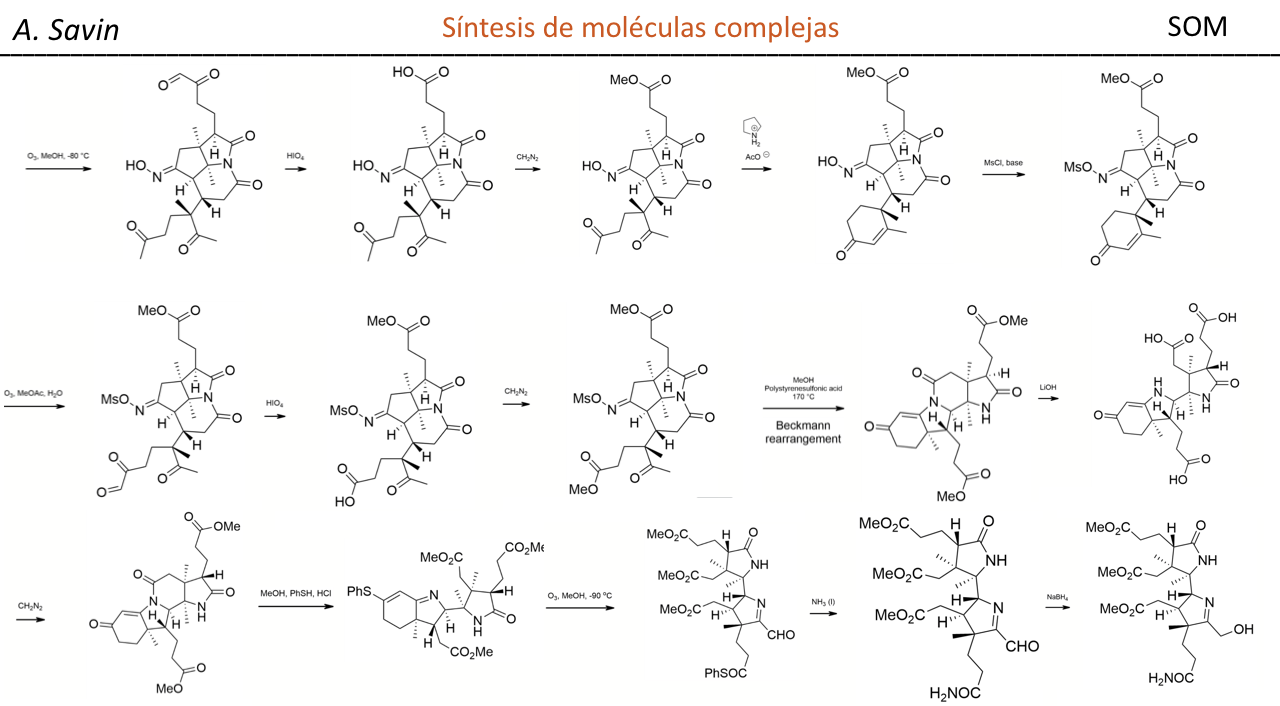
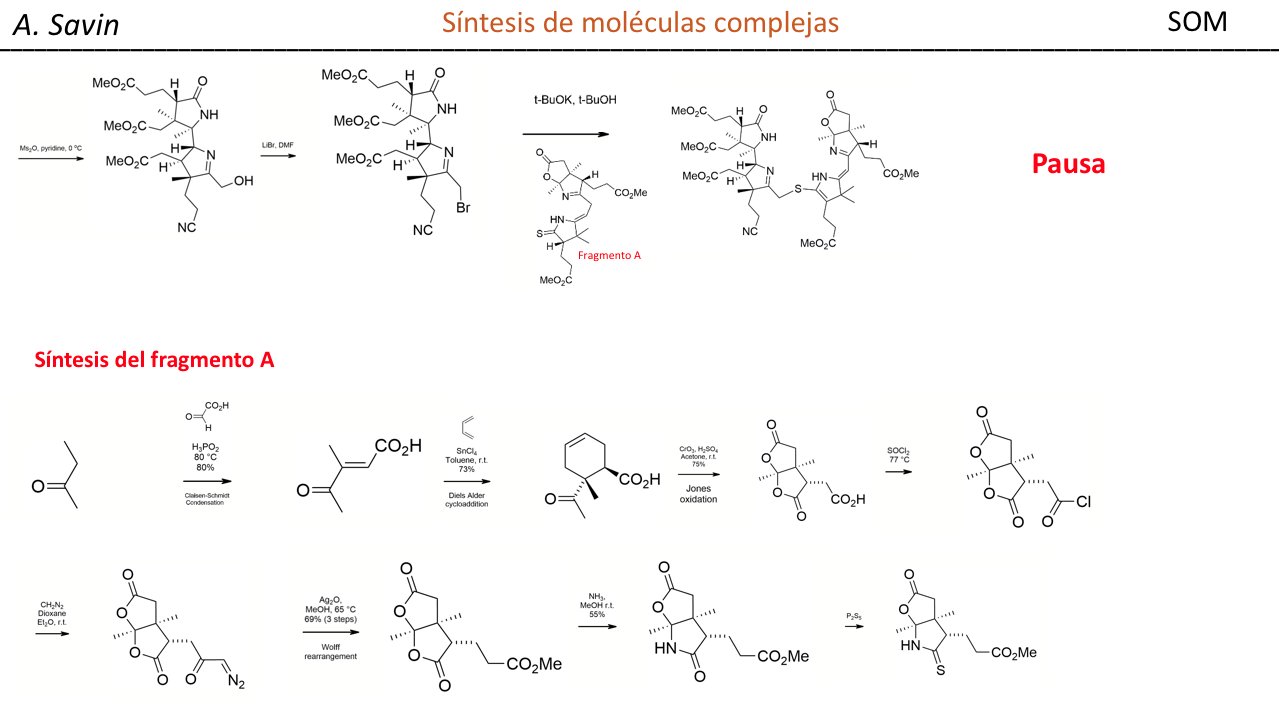
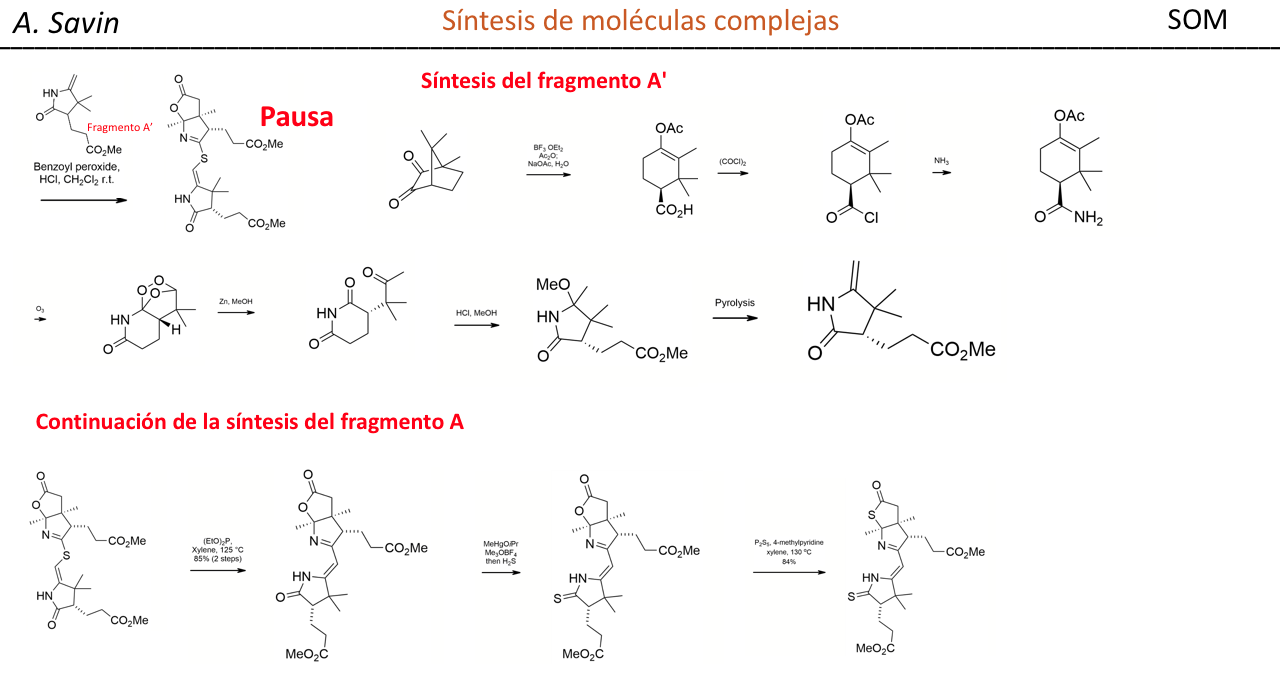
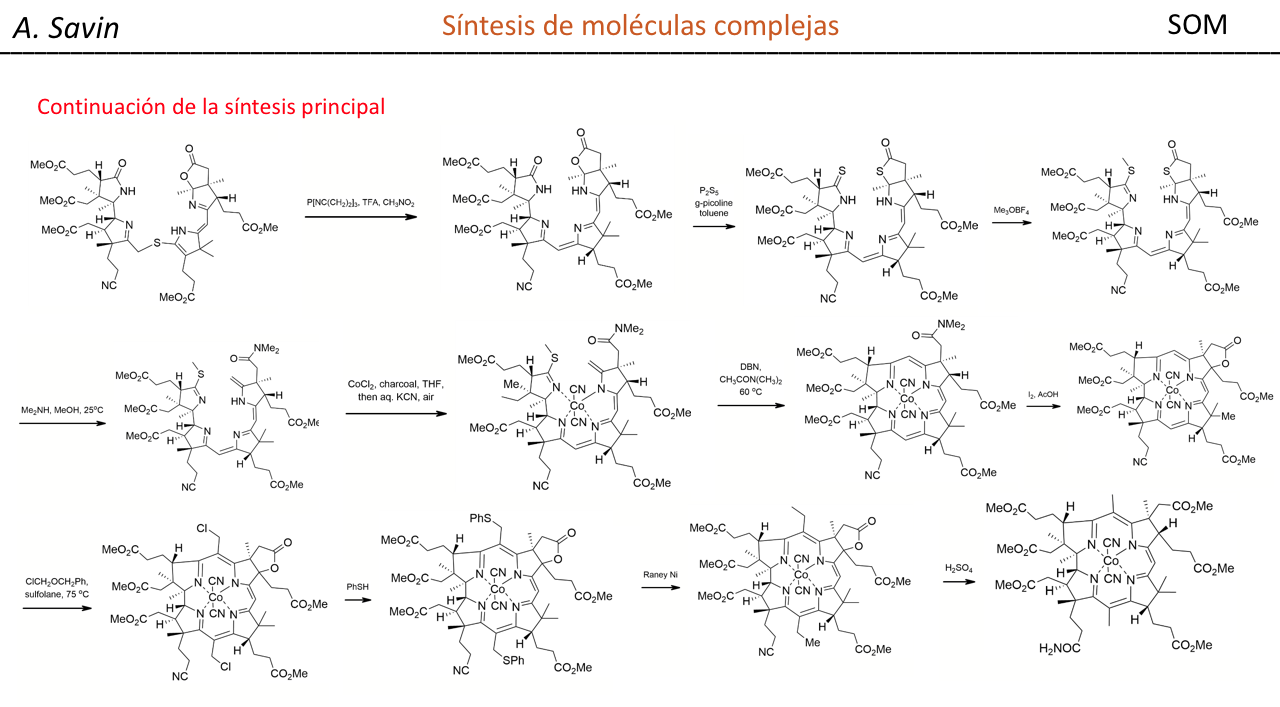
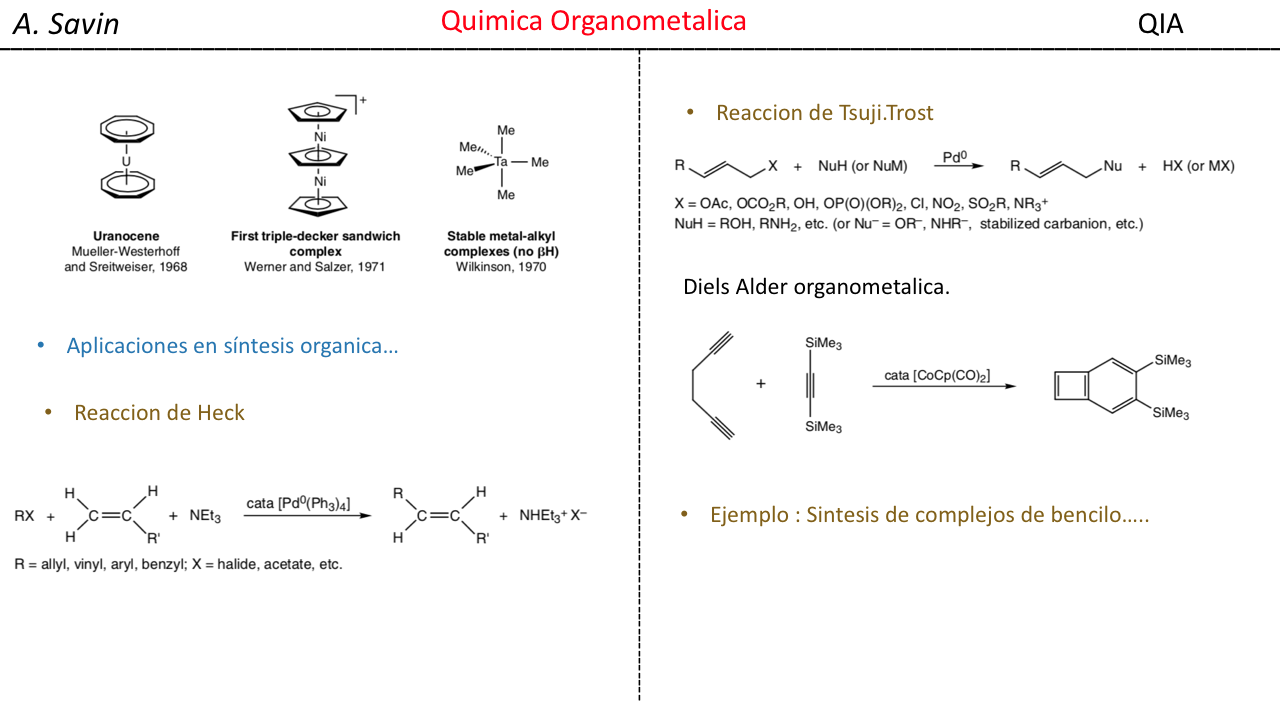
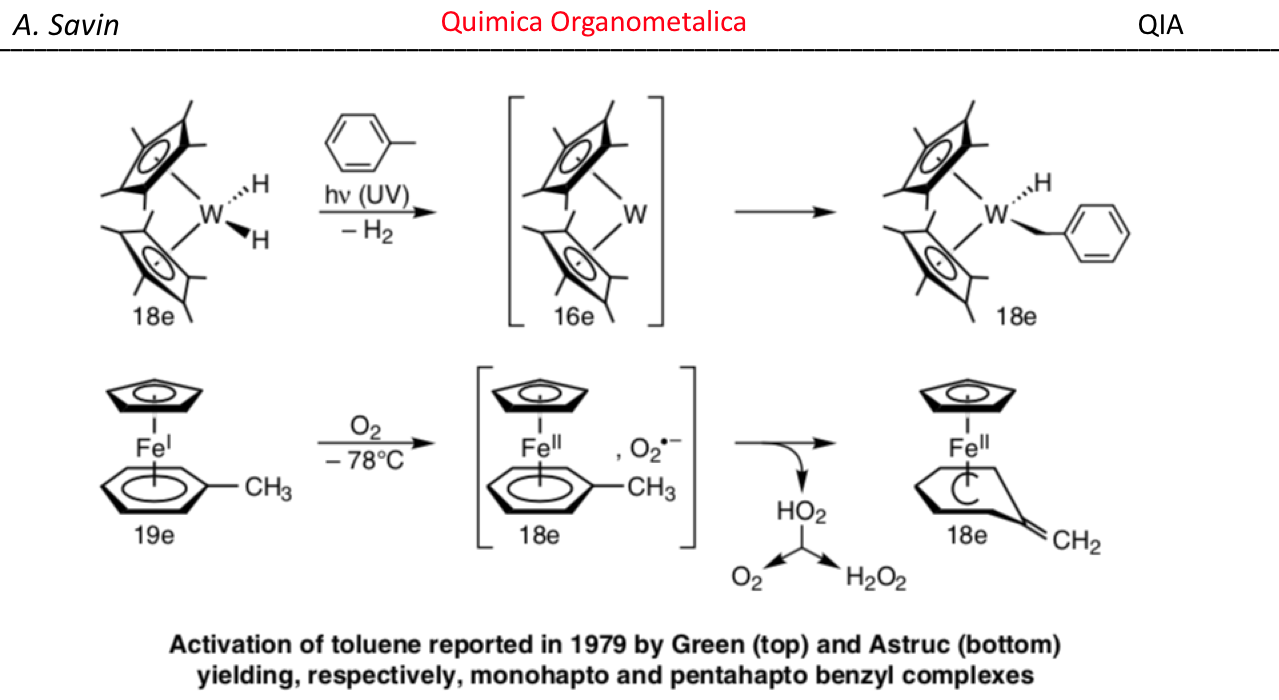
Alejandro Savin
- Detalles
- alejandro savin
- Visto: 15815
Efecto NOE
El efecto nuclear Overhauser ocurre cuando un protón, cuya proporción de núcleos con spin +1/2 y –1/2 está alterada (el cuel se consigue con una fuerte irradiación del protón) interactúa por acoplamiento dipolar con un protón vecino, alterando su proporción de núcleos con spin +1/2 y –1/2 y cambiando así la intensidad de la absorción. El desplazamiento químico no se altera.Se utiliza para detectar los protones que están cerca de otros. Es muy sensible a la distancia porque la interacción dipolar depende de r-6. El efecto NOE solo actúa a distancias menores a 5Å y no importa el número de enlaces químicos que separan a los dos protones.
Figura 1. Ejemplo de NOE
1- ¿De dónde viene el NOE?
Considere dos núcleos, I y S, que comparten un acoplamiento dipolar (a través del espacio). Estos acoplamientos dipolares dependen de la orientación relativa de I y S. En solución, el volteo molecular promedia estos acoplamientos, por lo que no aparecen en los espectros de RMN típicos. Sin embargo, la magnetización todavía se puede transferir entre ellos.
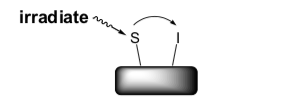
Figura 2. l y s representados.
Ahora si consideramos el efecto de perturbar las poblaciones de equilibrio del espín S de alguna manera en la intensidad de la señal del espín I. El NOE se define como:
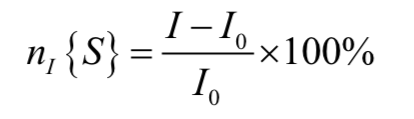
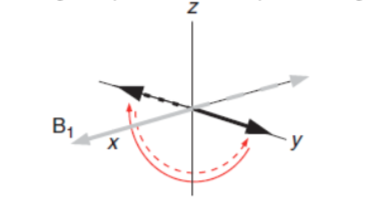
Para simplificar, supongamos que I y S no comparten un acoplamiento escalar (J). El experimento NOE más simple es el experimento de estado estacionario. Uno satura selectivamente el spin S, y luego aplica un pulso de 90 ° para observar el efecto que esto tiene sobre la población de spin en el spin I:
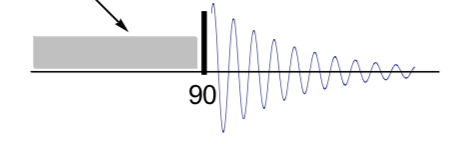
Figura 3. Pulso en RMN esquematizado a 90
Recordar que la saturación significa que la población de los niveles de energía alfa y beta en el giro S se iguala. En este caso, la cantidad de tiempo que se aplica la saturación es el tiempo de mezcla para el experimento; En un sentido general, los tiempos de mezcla más largos permiten más tiempo para la transferencia de magnetización.
 Ejemplo de NOE
Ejemplo de NOE
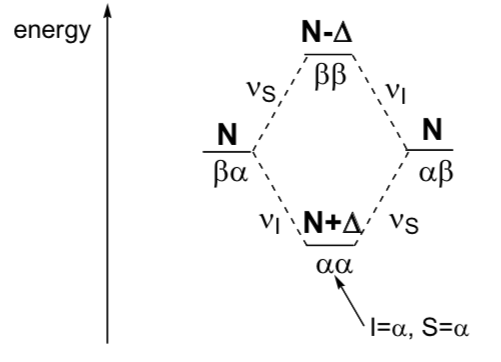
En el equilibrio, las diferencias de población entre los niveles de energía están determinadas por la distribución de Boltzmann. Llamemos a difference la diferencia de población para el spin I: en términos generales, el número de núcleos en exceso en el estado de energía más baja lower. Debido a que los cambios químicos son mucho más pequeños que la frecuencia de Larmor, el incremento también es la diferencia de población para el espín S (suponiendo, como estamos aquí, que I y S son ambos protones).
Para los núcleos 4N, el diagrama de energía es:
Aquí hay algunos espectros (arriba: NOE., Abajo: regular 1D):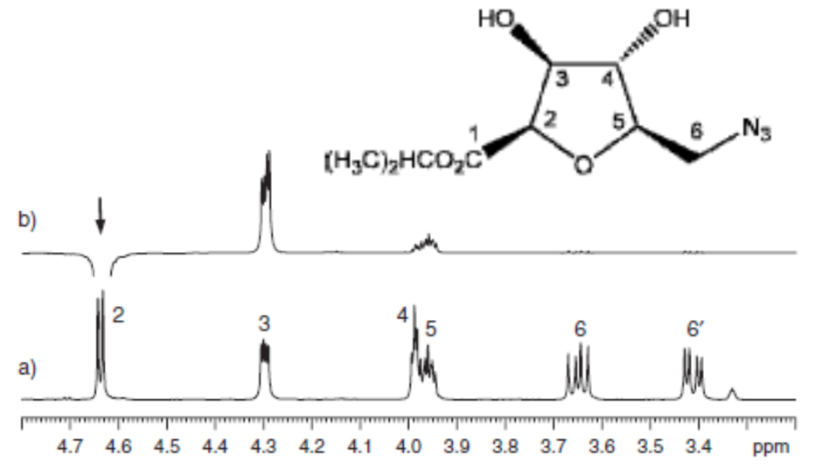
En el ejemplo anterior, parece que NOE se usó para asignar la geometría 2,5-cis en este anillo de cinco miembros. No conozco los detalles de este caso, pero en general, usar NOE para asignar la configuración relativa en anillos de cinco miembros es peligroso ya que son conformacionalmente flexibles.
 Referencias
1. Alonso, P. et al. quimicaCou..Ed. Mc Graw-Hill. 1990.2. Álvarez Jiménez, M. D. y Gómez del Río, M. I. Guía didacticaQuímica Analítica II. UNED. 1999.3. Arribas Jimeno Siro; Burriel Barcelo Fernando; Hernández Méndez Jesús; Lucena Conde Felipe. Química Analítica Cualitativa. ISBN: 8497321405. ISB. 2006.4. Ayres, Gilbert H. Análisis Químico Cuantitativo. Ediciones del Castillo, 4ta ed. ISBN: 8421902806. 1981.5. Bermejo Barrera. M del Pilar. Química analítica general, cuantitativa e instrumental. Editorial Paraninfo. 7ma Edición. ISBN: 8428318093. 1990.6. Blanco, M., Cerdá, V. y Sanz Medel, A., Espectroscopia Atómica Analítica, Publicaciones de la universidadAutónoma de Barcelona. 1990.7. Brode. R.W, Chemical spectroscopy, Nueva York 1952.8. Burriel, M.F., Lucena, C.F.Química Analítica Cuantitativa. Edición Revolucionaria. La Habana.1978.9. Burriel, F. Química Analítica Cualitativa. Editorial Paraninfo. ISBN: 8497321405. pp 1072. , 2003.
Referencias
1. Alonso, P. et al. quimicaCou..Ed. Mc Graw-Hill. 1990.2. Álvarez Jiménez, M. D. y Gómez del Río, M. I. Guía didacticaQuímica Analítica II. UNED. 1999.3. Arribas Jimeno Siro; Burriel Barcelo Fernando; Hernández Méndez Jesús; Lucena Conde Felipe. Química Analítica Cualitativa. ISBN: 8497321405. ISB. 2006.4. Ayres, Gilbert H. Análisis Químico Cuantitativo. Ediciones del Castillo, 4ta ed. ISBN: 8421902806. 1981.5. Bermejo Barrera. M del Pilar. Química analítica general, cuantitativa e instrumental. Editorial Paraninfo. 7ma Edición. ISBN: 8428318093. 1990.6. Blanco, M., Cerdá, V. y Sanz Medel, A., Espectroscopia Atómica Analítica, Publicaciones de la universidadAutónoma de Barcelona. 1990.7. Brode. R.W, Chemical spectroscopy, Nueva York 1952.8. Burriel, M.F., Lucena, C.F.Química Analítica Cuantitativa. Edición Revolucionaria. La Habana.1978.9. Burriel, F. Química Analítica Cualitativa. Editorial Paraninfo. ISBN: 8497321405. pp 1072. , 2003.
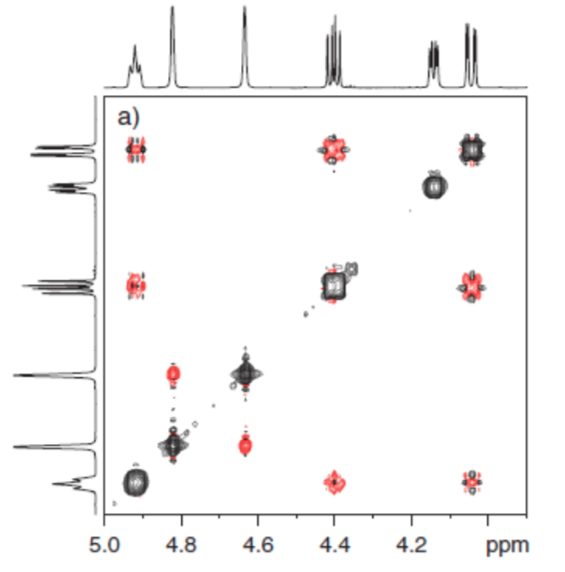
Como resultado, las secuencias de pulso ACOGEDOR y NOESY (y EXSY) son básicamente las mismas. En COSY, interesa la transferencia de magnetización a través de bonos; en NOESY, estamos interesados en la transferencia de magnetización a través del espacio. Las correlaciones ACOGIDAS surgen de la coherencia cuántica cero, que discutiremos en detalle más adelante en proximos blogs. Estos no pueden eliminarse mediante gradientes o ciclos de fase, pero se han desarrollado algunos métodos especiales como la "filtración z" para eliminarlos. (les contaré sobre eso más tarde en otro blog de estos contenidos de RMN.) El punto es que los cruces de COSY son fáciles de identificar porque tienen una fase "arriba-abajo". Son particularmente comunes cuando J es grande; por ejemplo, entre dos protones transdiaxiales en un ciclohexano.
Aquí hay un espectro NOESY sin ninguna eliminación de artefactos cuánticos cero:
Aquí, se usó un método sofisticado llamado "inversión de frecuencia barrida" para eliminar las señales COSY antifásicas: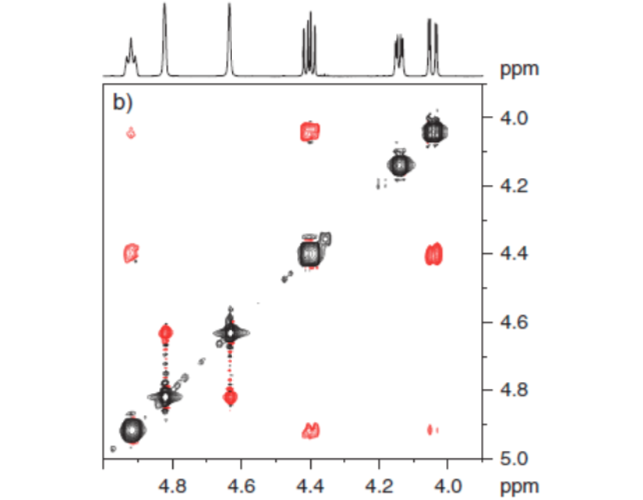
- Detalles
- Alejandro Savin.
- Visto: 72636
Espectroscopia de infrarrojo
La espectrometría del infrarrojo es sumamente útil para determinaciones cualitativas de compuestos orgánicos y para deducir estructuras moleculares a partir de sus grupos funcionales tanto de compuestos orgánicos como inorgánicos.
En el análisis cualitativo la espectroscopia de infrarrojo puede usarse para la identificación de sustancias puras o para la absorción, localización e identificación de impurezas.
Para localizar una impureza en una sustancia se hace una comparación en el espectro de las sustancia que se estudia y una muestra de la sustancia pura. Las impurezas causan bandas de absorción adicionales que aparecen en el espectro.
En el IR también están encontrando uso cada vez mayor en el análisis cuantitativo, el principal campo de aplicación de este tipo de análisis se halla en la cuantificación de contaminantes atmosféricos que provienen de procesos industriales.
Una parte del espectro electromagnético que se extiende desde 0.8 a 1000μm (que corresponde al número de onda comprendidos entre los 12800 y los 10 cm-1), se considera como la región del infrarrojo la cual está dividida en tres regiones llamadas:
a).- I.R. Cercano. b).- I.R. Fundamental ó Medio c).- I.R. Lejano
Cada tipo de enlace absorbe radiación infrarroja a una frecuencia distinta, lo que permite determinar que tipo de grupos funcionales posee la molécula en estudio. Los espectrofotómetros de infrarrojo trabajan en el infrarrojo medio y hacen un barrido desde los 4000 cm−1 hasta los 400 cm−1
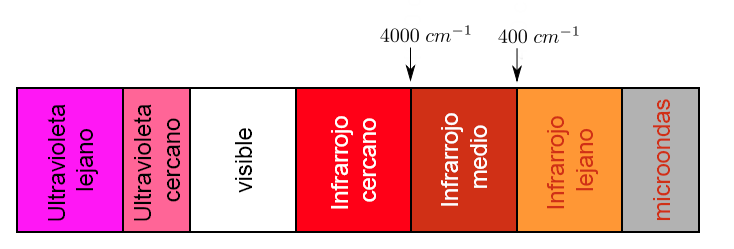
Sin embargo la región de importancia analítica es la región del I.R. Fundamental ya que la mayoría de los instrumentos infrarrojos cubren ésta región.
La mayoría de los materiales orgánicos e inorgánicos demuestran absorción y el espectro es originado principalmente por el alargamiento vibracional y flexión dentro de la molécula. El espectro infrarrojo es una de las propiedades más características de un compuesto ya que no existen dos espectros iguales para dos compuestos diferentes, es como una huella dactilar.
Dentro de la región del I.R. Fundamental existen dos regiones, una de ellas es la llamada de
los grupos funcionales de 4000cm-1 a 1300 cm-1, y la región dactilar de 1300 cm-1 a 670cm-1 .
En la región de los grupos funcionales la posición del pico de absorción es mayor o menor dependiendo solamente del grupo funcional donde llega la absorción y no de la estructura molecular completa. La posición de los picos en la región dactilar son dependientes de la estructura molecular completa.
Características que debe tener una vibración para que produzca banda de absorción:
La radiación incidente debe tener una frecuencia igual a la frecuencia de la vibración que va a producir.
Que la vibración resultante produzca cambio en el momento dipolar, o sea que la vibraciónno absorberá radiación infrarroja, si no hay cambio en el momento dipolar se llama vibración inactiva y serán activas cuando haya dicho cambio en el momento dipolar.
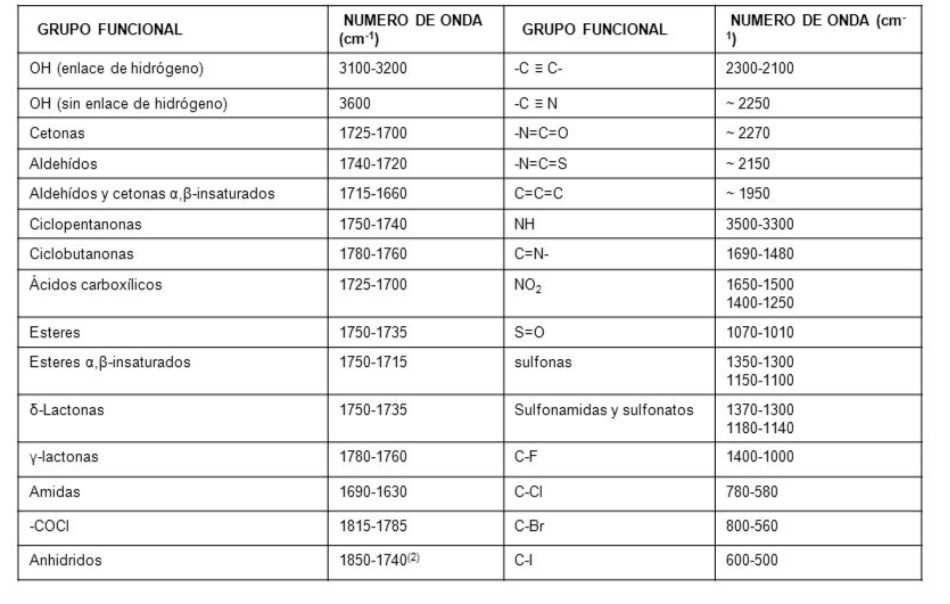
Tradicionalmente, en el eje x de los espectros de infrarrojo se emplea el número de ondas (ν¯, léase "nu barra"') y se define como el inverso de la longitud de onda en cm. ν¯=1λ. En el eje y se representa el porcentaje de radiación transmitida (transmitancia) que se representa por %T. A continuación, se muestra la forma que presenta el espectro de infrarrojos del hexano.
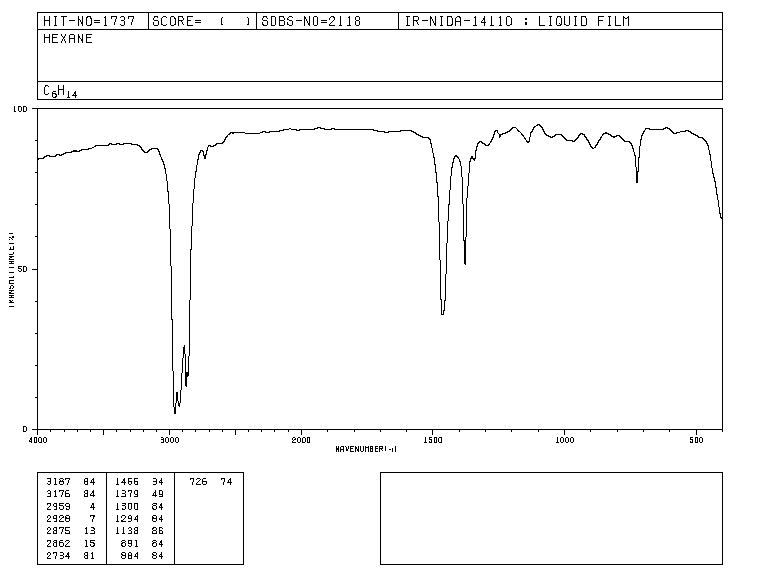
Las bandas representan zonas donde los enlaces de la molécula absorben radiación infrarroja. En las bandas la transmitancia es pequeña y la absorbancia grande.
Tipos de vibracion
Vibración de tensión (stretching). Los átomos unidos por enlaces simples, dobles o triples se acercan y alejan siguiendo la dirección del enlace, igual que oscilan dos masas unidas por un muelle.
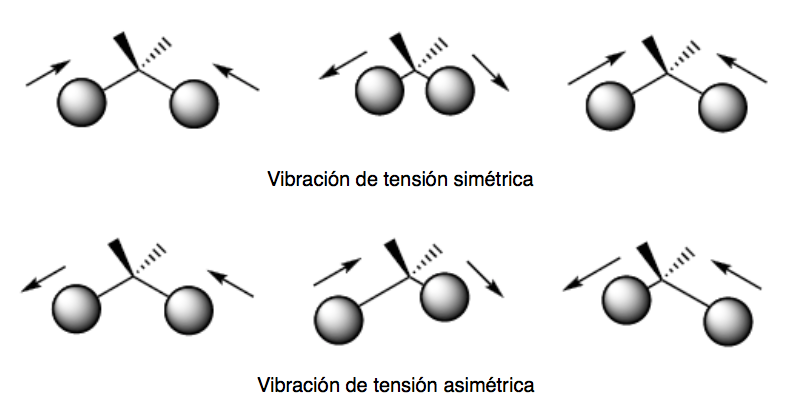
Hay dos modos de vibración de tensión: simétrica y asimétrica
vibración de flexión (bending). Los átomos vibran de modo que varían los ángulos, pero no las longitudes de enlace. Hay cuatro modos de vibraciones de flexión: tijera (scissoring), balanceo (rocking), cabeceo (wagging) y torsión (twisting)
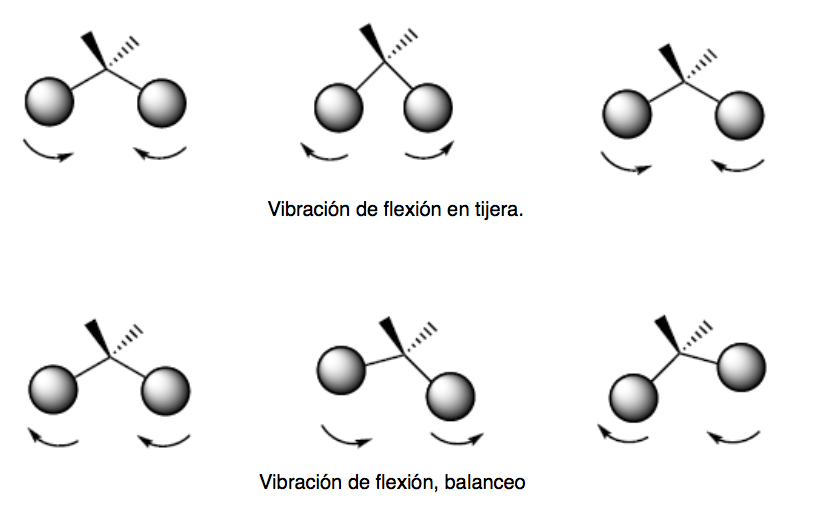
Estos dos modos de vibración tienen lugar en el plano que contiene los tres átomos que participan en la vibración
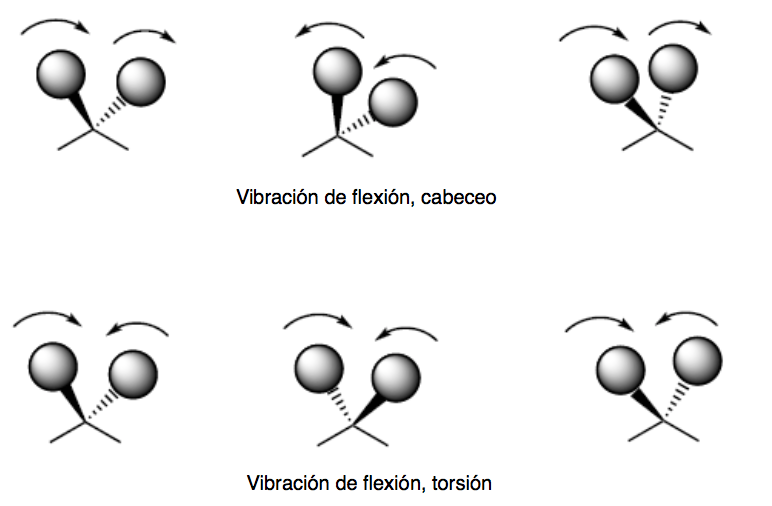
Los modos de vibración de cabeceo y torsión tienen lugar fuera del plano (Out of plane) y suelen representarse por Oop.
Oscilador armonico cuantico
Las vibraciones moleculares pueden estudiarse con el modelo del oscilador armónico cuántico. La energía viene dada por:
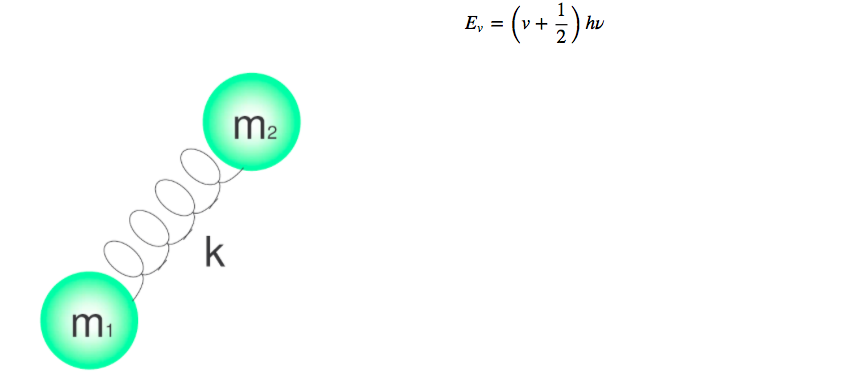
Los distintos niveles de energía vienen dados por el número cuántico v, que toma valores 0.1.2.3.4.....
h es la constante de Planck y ν la frecuencia del oscilador que viene dada por la expresión:
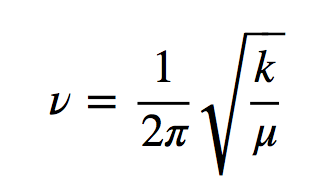
Donde k es la constante de fuerza del muelle y μ la masa reducida del sistema
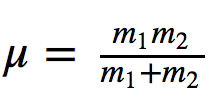
Dividiendo la frecuencia entre la velocidad de la luz se obtiene número de ondas ν¯
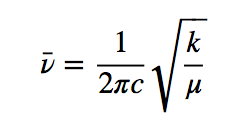
La ecuación anterior indica que masas reducidas pequeñas (átomos de poca masa) y constantes de fuerza altas (enlaces fuertes) conducen a frecuencias altas. En estas condicionees las bandas de absorción salen a numeros de onda altos.
Como puede observarse en el gráfico las frecuencias altas dan lugar a un mayor espaciado entre los niveles energéticos.
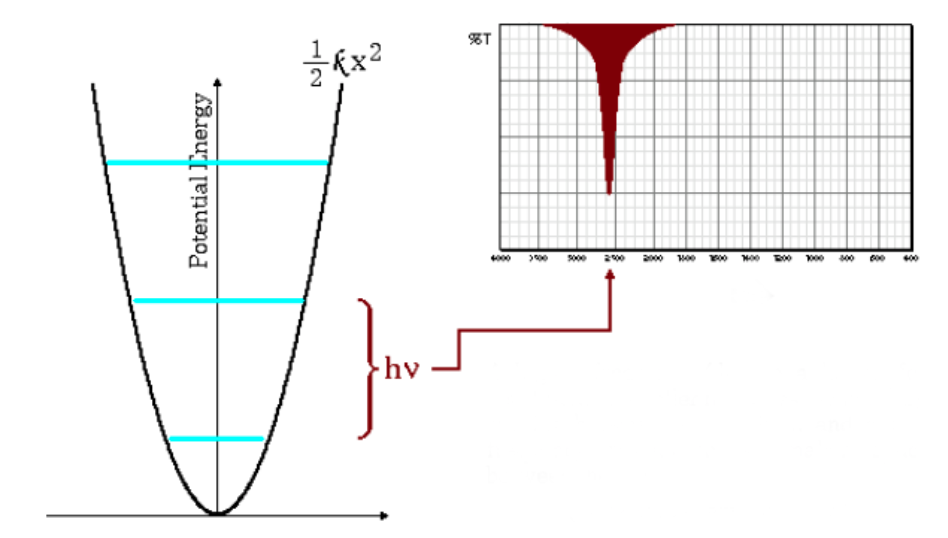
Frecuencias de absorción bajas
La ecuación antetior tambien indica que masas reducidas grandes y constantes de fuerza pequeñas (enlaces débiles) conducen a frecuencias bajas. En estas condicionees las bandas de absorción salen a numeros de onda bajos.
Como puede observarse en el gráfico las frecuencias bajas dan lugar a un menor espaciado entre los niveles energéticos.
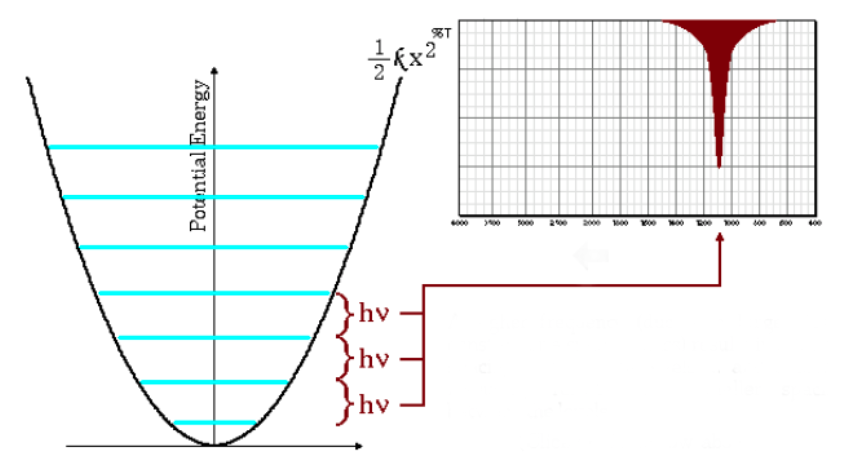
Principales vibraciones moleculares
Espectro IR
En un espectro de infrarrojos se representa la frecuencia (en número de onda) frente al porcentaje de luz transmitida (transmitancia). El porcentaje de transmitancia se define como el cociente entre la intensidad de la luz transmitida a través de la muestra, IM, y la intensidad de la luz del haz de referencia IR multiplicado por 100.
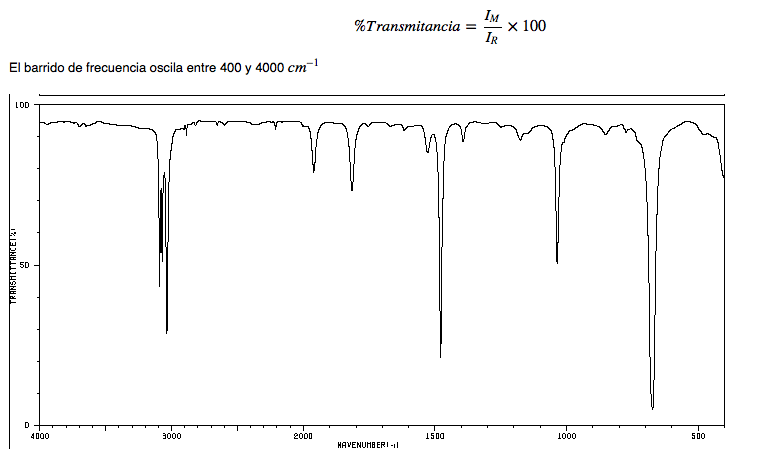
Espectro IR de alcanos
• Tensión C-H: los alcanos presentan vibraciones de tensión C-H ligeramente por debajo de 3000 cm−1
• Flexión C-H: los CH2 de la cadena presentan vibraciones de flexión (tijera) a 1465 cm−1, mientras que los metilos producen una banda a 1375 cm−1debida a la vibración de flexión simétrica y otra a 1450 cm−1 debida a la vibración de flexión asimétrica. Todas las bandas de flexión son de intensidad media.
Obsérvese que la banda de flexión asimétrica del metilo solapa con la de flexión en tijera del CH2.
Espectro IR del hexano
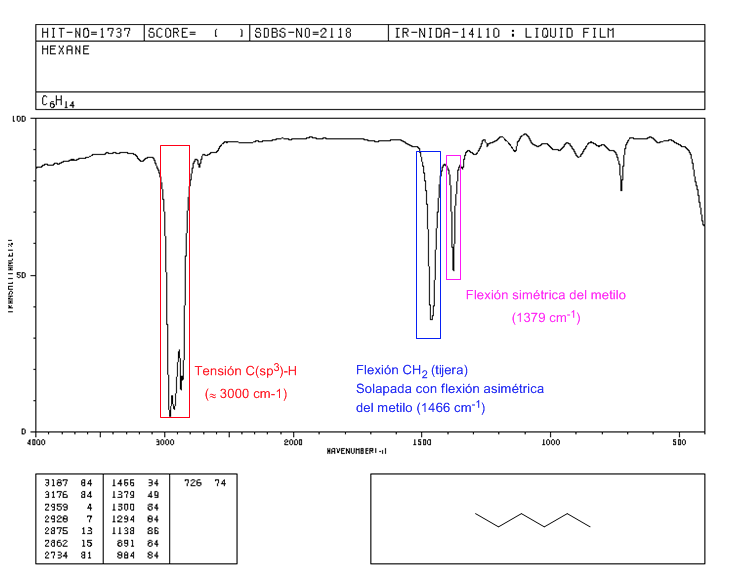
Espectro del 2,2-dimetilbutano
La presencia del grupo tert-butilo produce el desdoblamiento de la banda de flexión simétrica en dos bandas a 1390 y 1370 cm−1. La banda a 1390 tiene la mitad de intensidad que la de 1370.
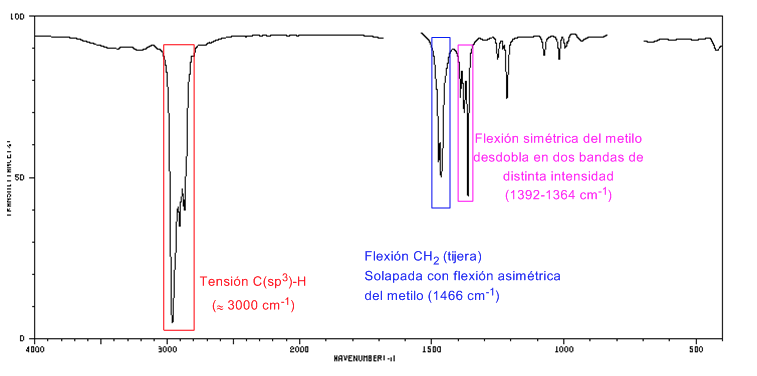
Espectro IR de cicloalcanos
Los cicloalcanos tienen un espectro de IR muy similar a los alcanos con banda de tensión C-H ligeramente por debajo de 3000 cm−1 y banda de flexión C-H en tijera para los CH2 a 1465 cm−1. La principal diferencia con los alcanos es la ausencia de la banda de tensión simétrica del metilo.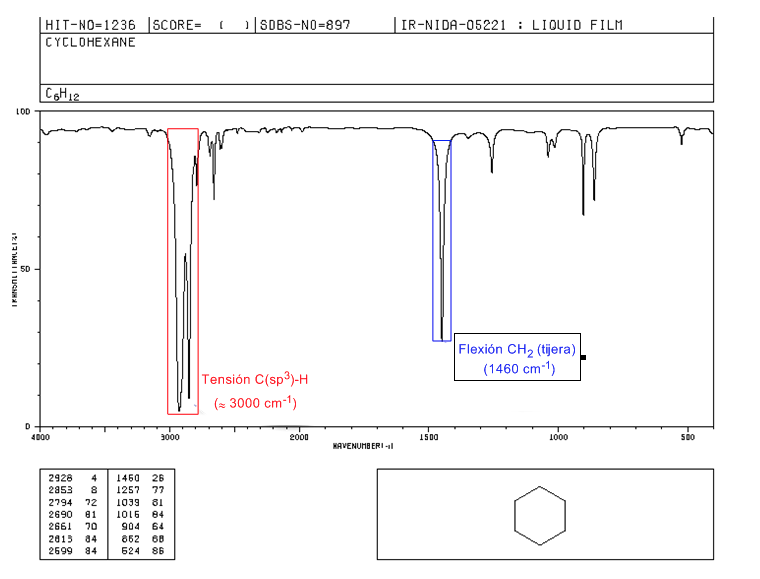
Obsérvese la ausencia de la banda de flexión simétrica del metilo que presentan los alcanos a 1375 cm−1.
Espectro IR de alquenos
• Tensión C(sp2)-H: 3100 -3000 cm-1
• Tensión C=C: 1600 cm-1
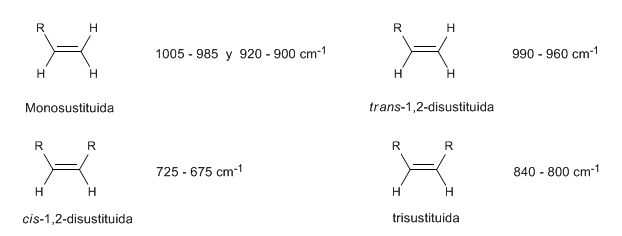
• Flexión fuera del plano (oop) del enlace C=C-H: 1000 - 650 cm-1. Este tipo de banda permite conocer el grado de sustitución del alqueno.
Espectro del 1-penteno
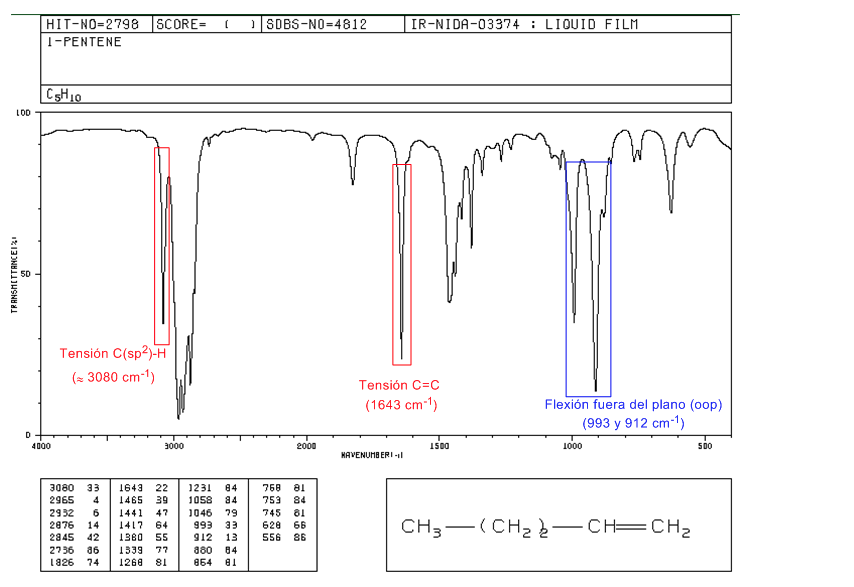
En los alquenos monosustituidos, como el 1-penteno, las flexiones C-H fuera del plano producen dos bandas situadas en 1005-985 y 920-900 cm−1.
Estereoquimica y espectroscopia IR de alquenos
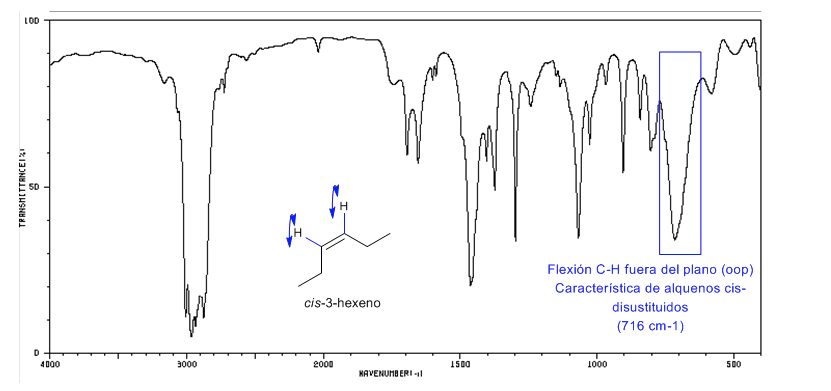
Los alquenos cis-disustituidos presenan una banda de flexión C-H fuera del plano que permite distinguirlos. Esta banda aparece entre 725-675 cm−1
Espectro del Cis-3-hexeno
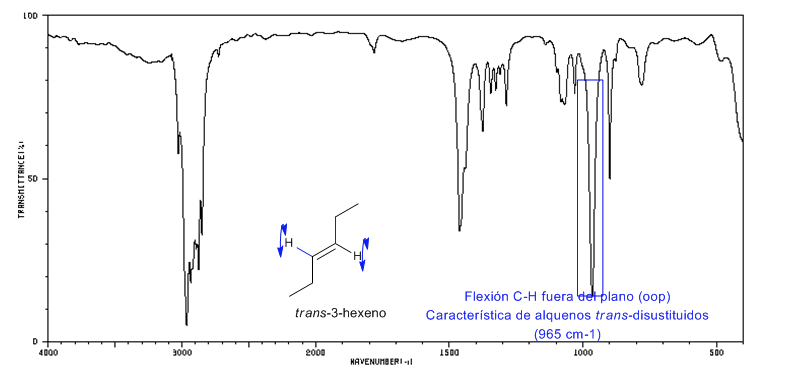
Espectro del Trans-3-hexeno
Los alquenos trans-disustituidos presentan una banda de absorción fuerte entre 980-965 cm−1 que permite identificarlos. Obsérvese la ausencia total de la banda de tensión C=C a 1600 cm−1 debido a la falta de polaridad.
Espectro del Metilenciclopentano
El metilenciclopentano constituye un ejemplo de olefina 1,1-disustituida y presenta una banda de flexión C-H fuera del plano muy intensa, localizada entre 900-880 cm−1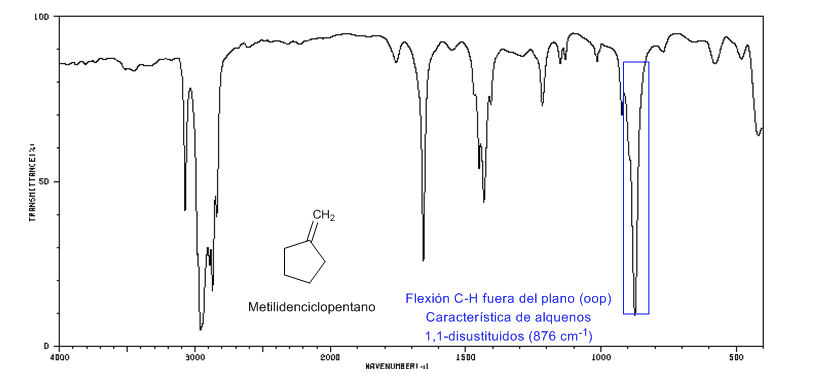
Resumen de flexiones C-H fuera del plano (oop)
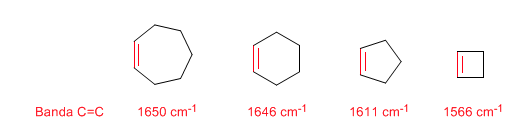
Tensión de anillo: enlaces C=C endocíclicos
Al disminuir el tamaño del anillo la banda de tensión de los enlaces C=C se desplaza hacia menor número de ondas. La excepción del ciclopropeno se atribuye al acoplamiento entre las vibraciones de tensión de los enlaces C=C y C-C. Este acoplamiento no se produce en el ciclobuteno debido a que los enlaces C=C y C-C se encuentran perpendiculares entre sí.
 Espectro IR del ciclopenteno
Espectro IR del ciclopenteno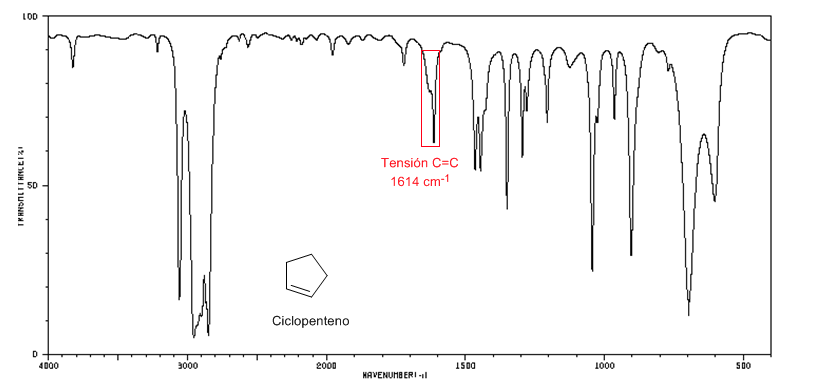
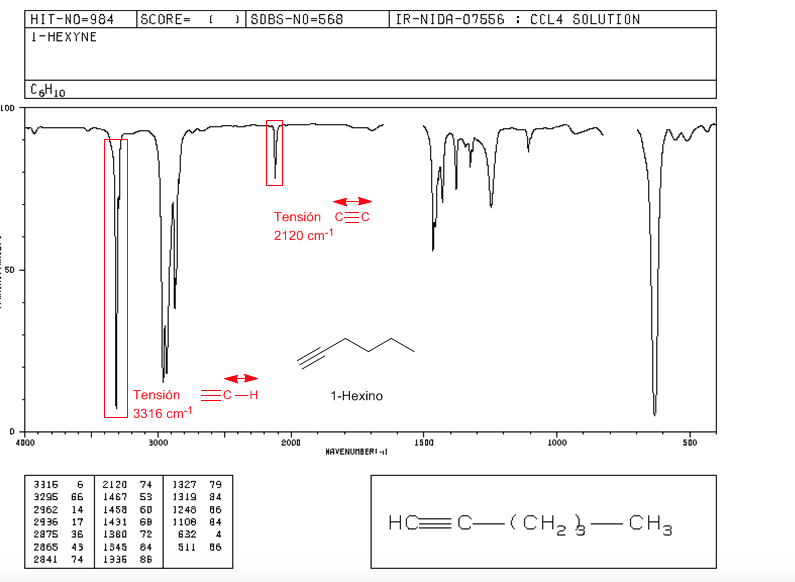
Espectros IR de alquinos
Tensión ≡C−H: 3300 cm−1
Tensión −C≡C−: 2150 cm−1. Los alquinos simétricos no presentan esta banda, siendo muy débil en los internos. La conjugación baja ligeramente el valor.
Espectro del 1-hexino
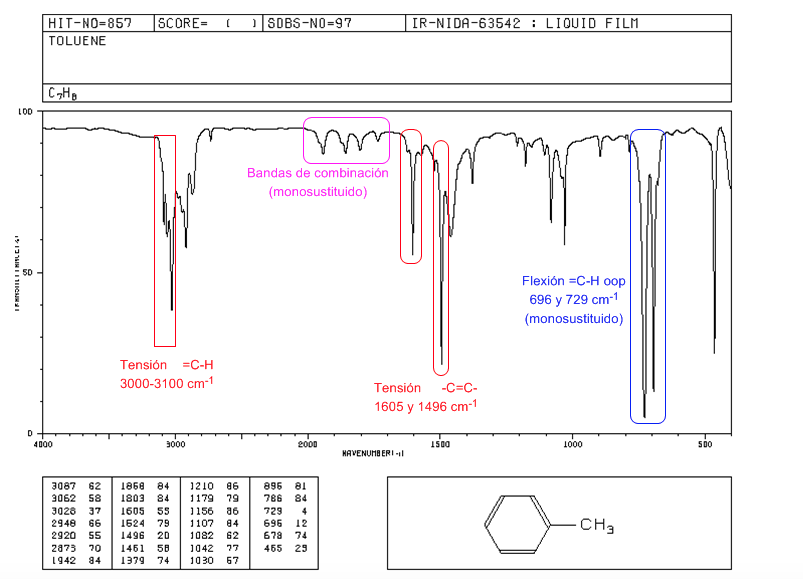
Espectro IR de aromaticos
Tensión =C-H: 3100 cm-1
Tensión -C=C-: 1600 y 1475 cm-1
Flexión =C-H fuera del plano: 900-690 cm-1
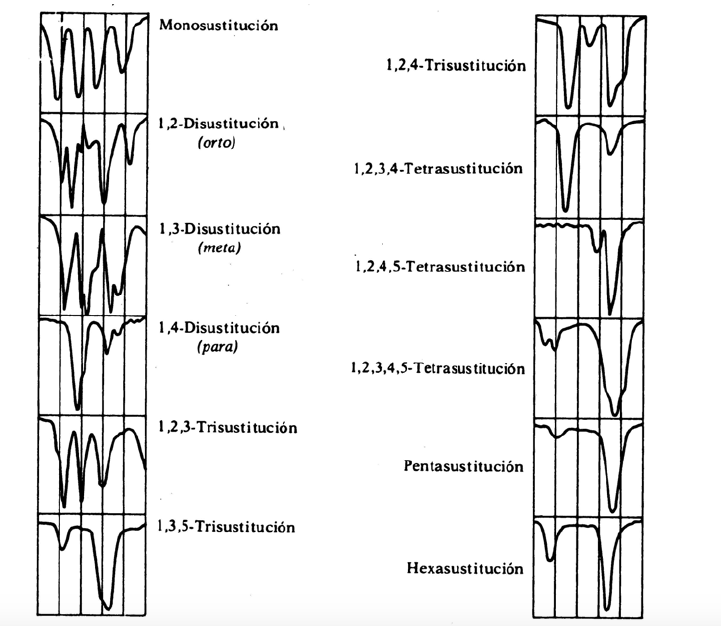
Las vibraciones oop junto con los sobretonos y bandas de combinación que aparecen entre 2000 y 1667 cm-1 permiten conocer el grado de sustitución del benceno.
Bandas de sustitucion
![]()
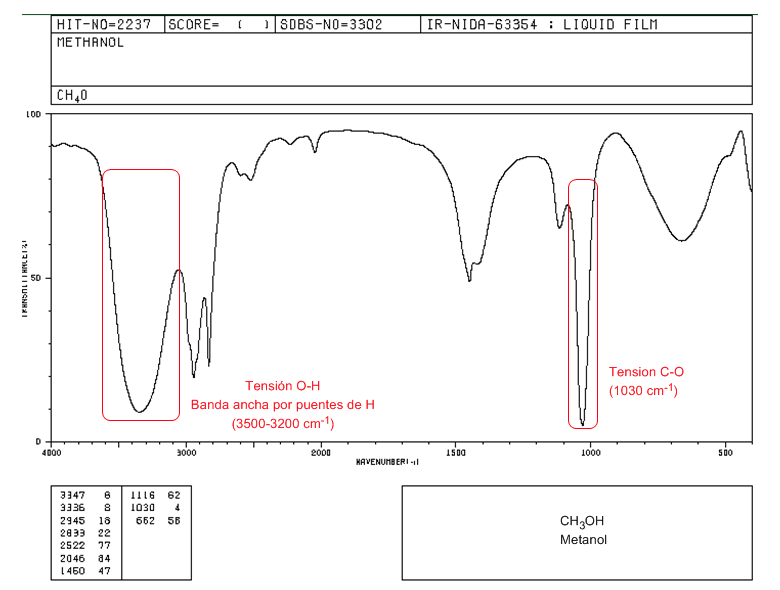
Espectros de alcoholes y fenoles
Tensión O-H: Banda ancha desde 3500 a 3200 cm-1. En ausencia de puentes de hidrógeno aparece como un pico agudo a 3650-3600 cm-1.
Tensión C-O: Banda comprendida entre 1250-1000 cm-1. Permite distinguir entre alcoholes primarios (1050 cm-1), secundarios (1100 cm-1), terciarios (1150 cm-1) y fenoles (1220 cm-1).
Espectro IR de un alcohol primario (metanol)
En el espectro del metanol podemos observar la banda de tensión O-H, muy ancha, por formación de puentes de hidrógeno. La banda de tensión C-O sale a número de ondas bajo (1030) por tratarse de un alcohol sin sustituyentes.
Espectro IR de alcoholes secundarios (2-pentanol)
Obsérvese el desplazamiento de la banda C-O hacia mayor número de ondas con respecto al metanol.
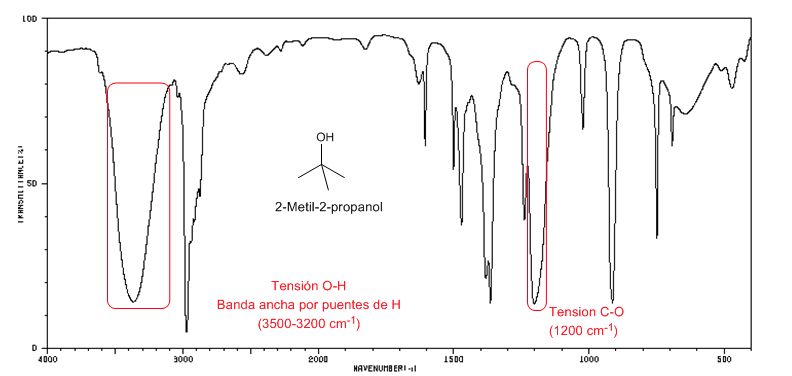
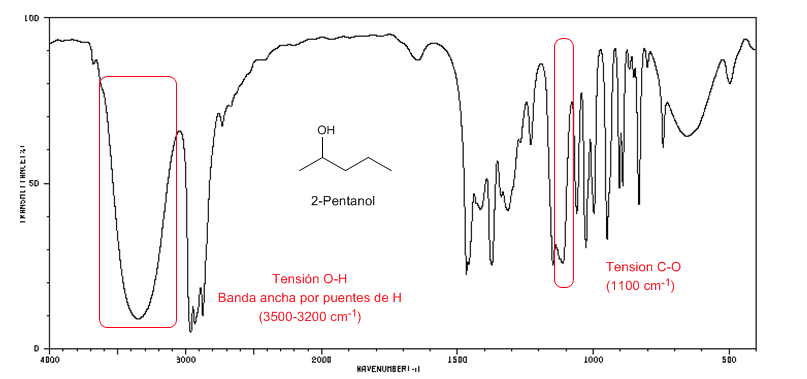 Espectro IR de alcoholes terciarios ( 2-metil-2-propanol)
Espectro IR de alcoholes terciarios ( 2-metil-2-propanol)Los alcoholes terciarios tienen la banda C-O desplazada a frecuencias mayores que los alcoholes primarios y secundarios.
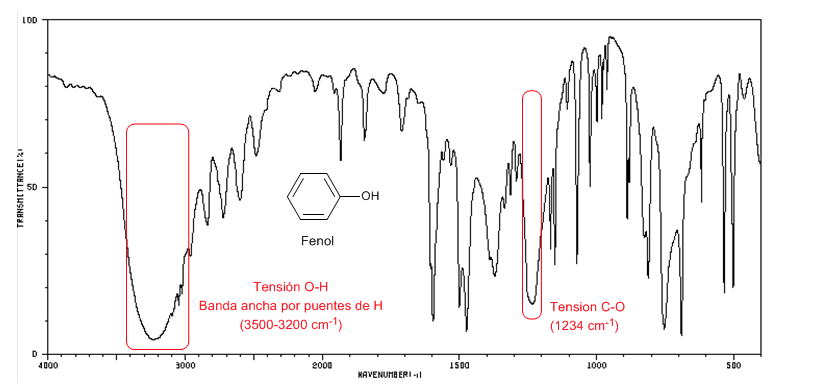
Espectro IR de fenoles ( fenol)
El fenol presenta una banda de absorción C-O por encima de 1200 cm-1
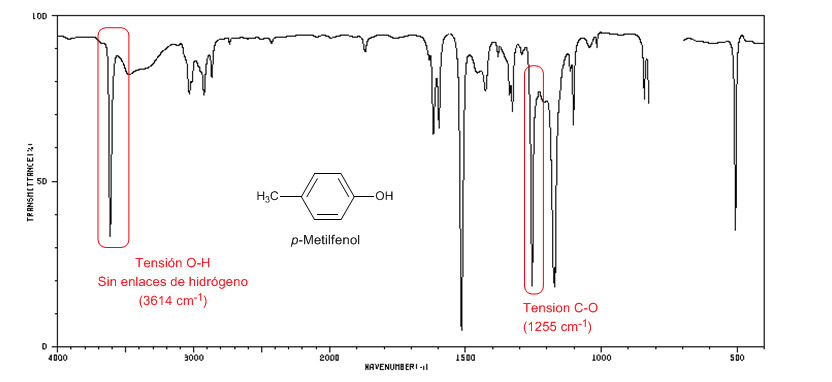
Espectro IR de fenoles en ausencia de puentes de hidrogeno (P-metil-fenol) en CCl4
El siguiente espectro muestra la banda de tensión O-H en ausencia de puentes de hidrógeno.
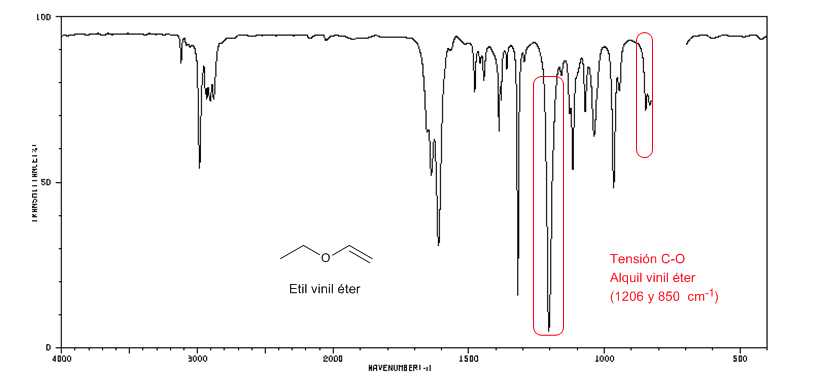
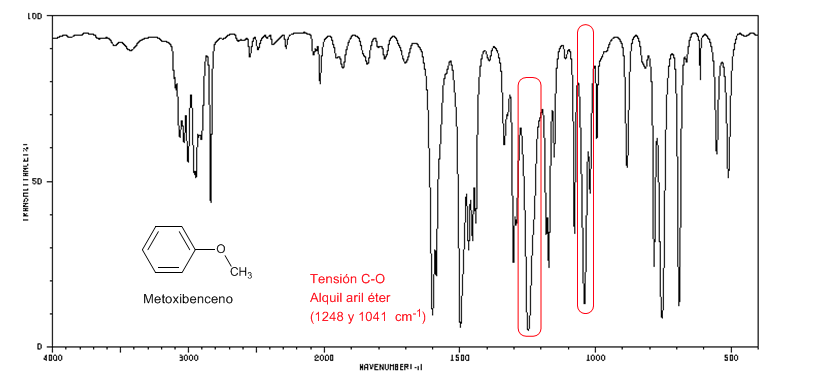
Espectro de eteres
Tensión C-O: 1300-1100 cm-1. Los dialquil éteres (R-O-R) presentan una banda a 1120 cm-1
Los alquil vinil éteres (CH2=CH-O-R) presentan dos bandas a 1220 y 850 cm-1. Esta última muy débil.
Los aril alquil éteres (Ar-O-R) presentan dos bandas a 1250 y 1040 cm-1
Espectro IR del 1metoxihexano
Espectro IR del etil-vinil-eter
El etil vinil éter presenta dos bandas a 1220 y 850 cm-1.Esta última muy débil.
Espectro IR del metoxibenceno
El metoxibenceno presenta dos bandas a 1250 y 1040 cm-1
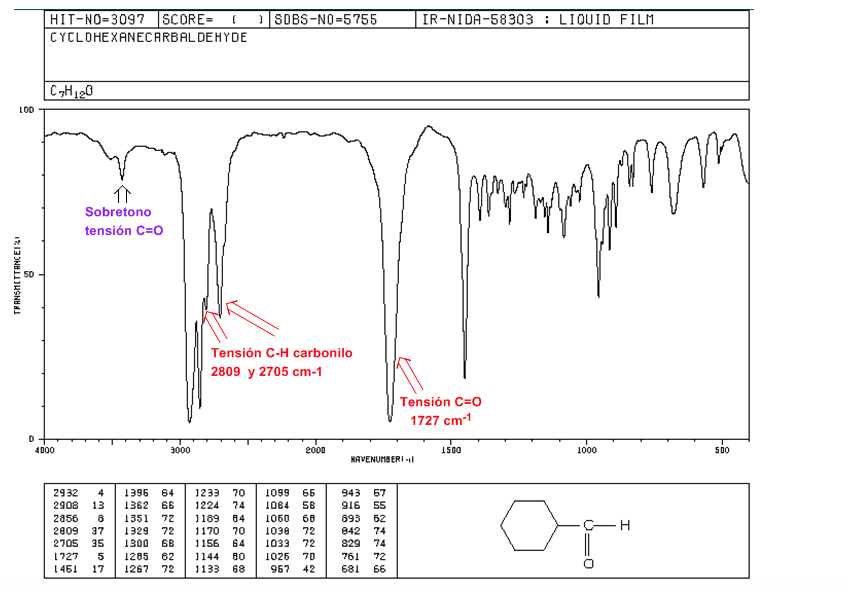
Espectro IR de aldehidos y cetonas
Aldehidos
Tensión C=O: 1725 cm-1
Tensión C-H carbonilo: dos bandas débiles a 2850 y 2750 cm-1. La banda a 2850 suele solaparse con la de tensión C(sp3)-H
Sobretono de Tensión C=O sobre 3500 cm-1.
Espectro IR del ciclohexanocarbaldehido
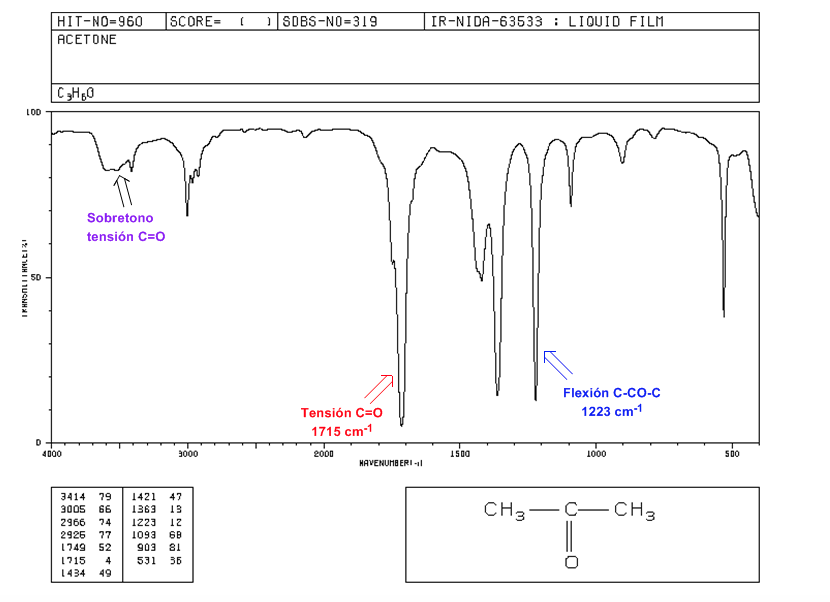
Cetonas
Tensión C=O: Banda intensa a 1715 cm-1.
Flexión C-CO-C: 1300 - 1100 cm-1.
Sobretono de tensión C=O: desde 3500 a 3350 cm-1.
Espectro del la propanona
Espectro IR de acidos carboxilicos y derivados
Acidos carboxilicos
Tensión O-H: Desde 3400 a 2400 cm-1. Muy ancha debido a la formación de puentes de hidrógeno.
Tensión C=O: 1730-1700 cm-1
Tensión C-O: 1320-1200 cm-1
Flexión C-O-H (oop): Banda en forma de campana a 900 cm-1
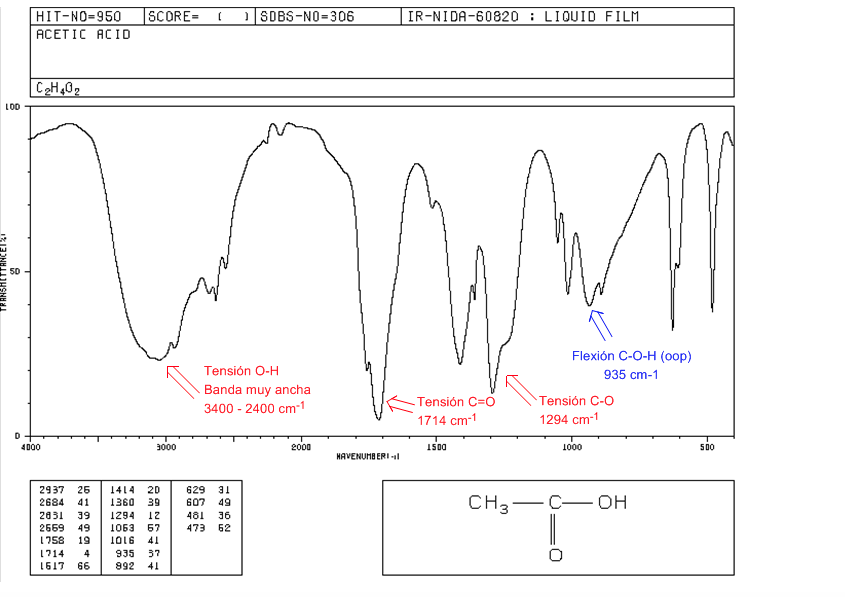
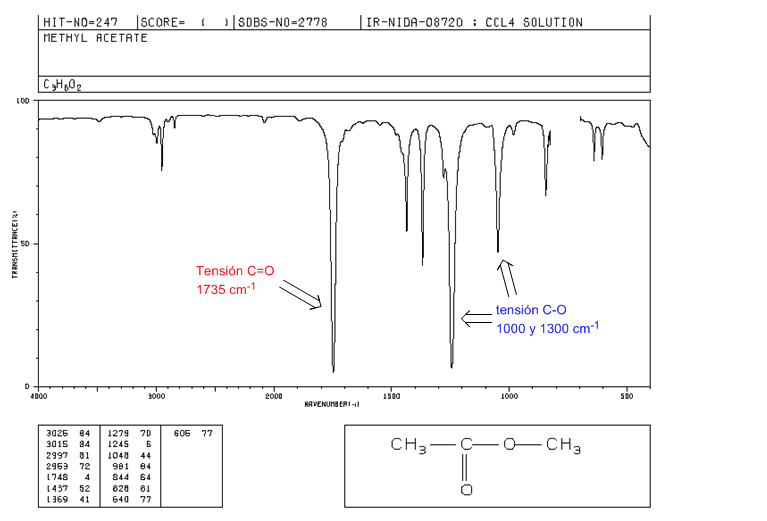
Esteres
Tensión C=O a 1735 cm-1. Si existen dobles enlaces conjugados con el carbonilo la banda se desplaza a valores más bajos. Cuando el doble enlace se encuentra sobre el grupo alcoxi (-OR) del éster se observa un desplazamiento hacia valores más altos.
Tensión C-O: 2 bandas a 1300 y 1000 cm-1. Siendo más ancha e intensa la obsevada a 1300.
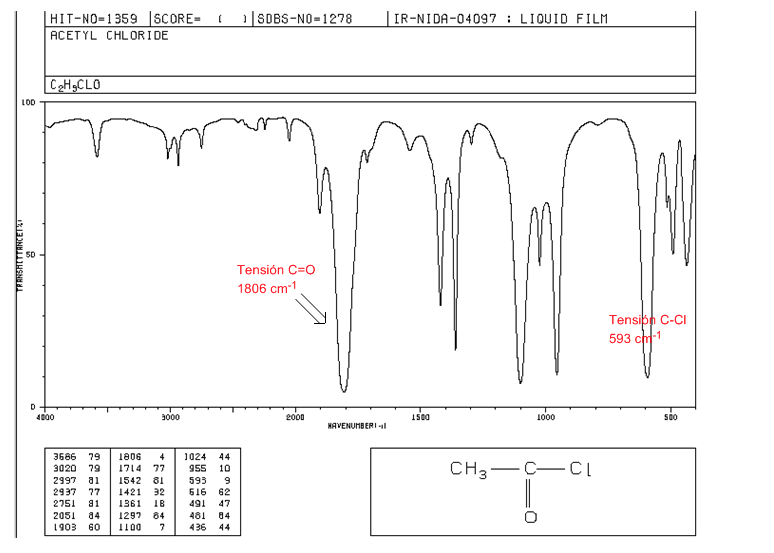
Haluros de acido
Tensión C=O: 1810 - 1775 cm-1
Tensión C-Cl: banda intensa 730 - 550 cm-1
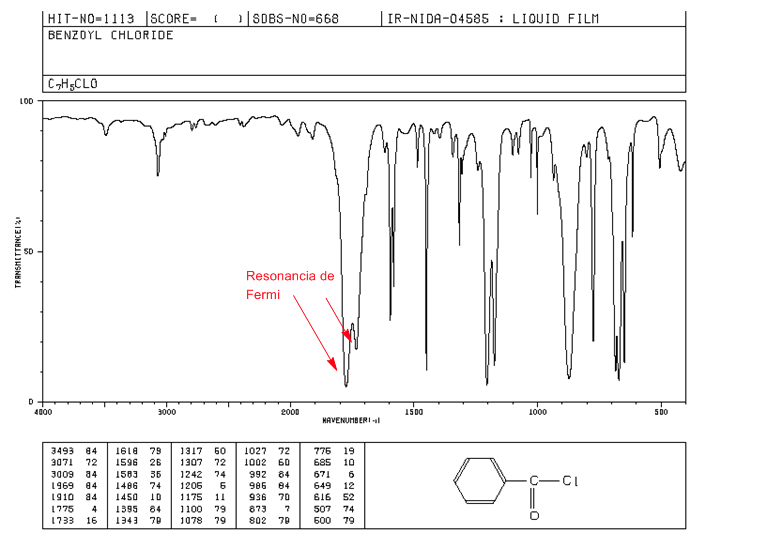
Resonancia de Fermi
Sobretonos, son transiciones vibracionales desde el estado fundamental a estados excitados superiores. Las frecuencias de absorción son siendo la frecuencia de la absorción fundamental. Resonancia de Fermi, resultan del acoplamiento de una banda de absorción fundamental con un sobretono o una banda de combinación.
Los haluros de alcanoílo aromáticos presentan dos bandas de tensión C=O por resonancia de Fermi.
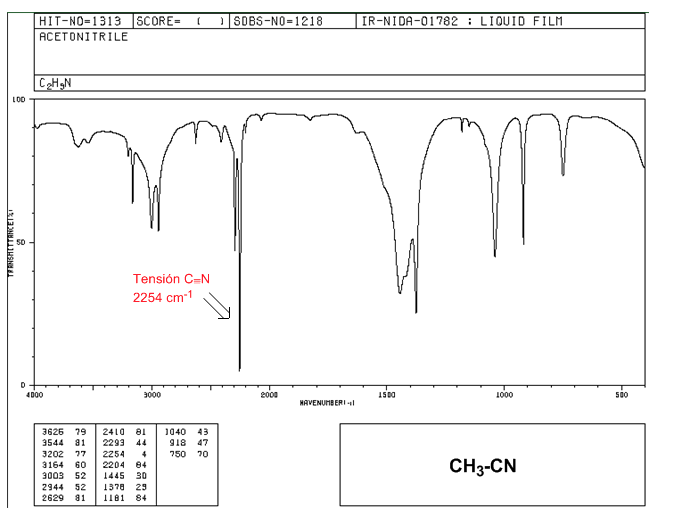
 Nitrilos
NitrilosTensión C≡N: banda muy fina a 2250 cm-1
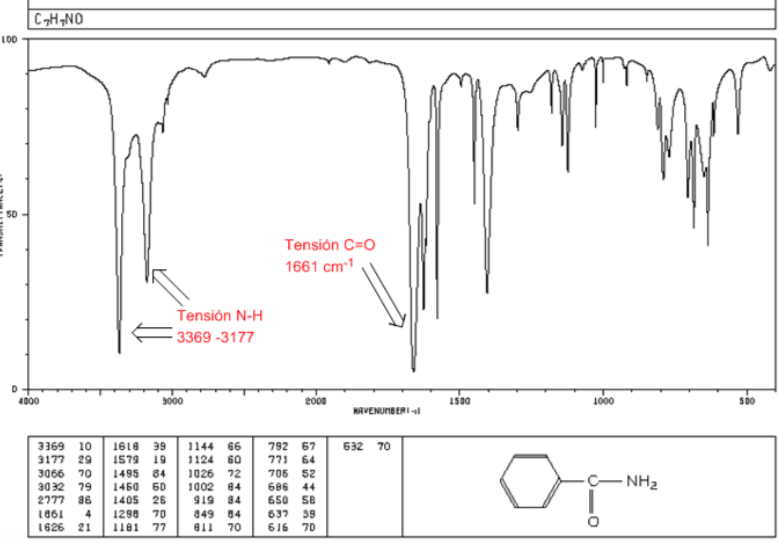
Amidas
Tensión C=O: 1680 - 1630 cm-1
Tensión N-H: Entre 3350 y 3180 cm-1. Las amidas primarias presentan dos bandas, mientras que las secundarias tienen una sóla banda.
Flexión N-H: 1640 - 1550 cm-1
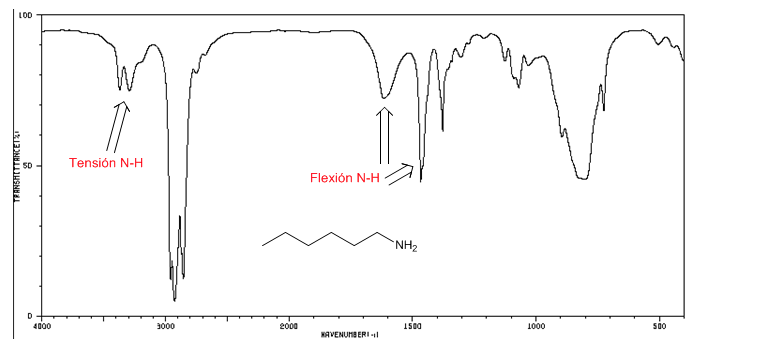
Espectro IR de aminas
Tensión N-H: entre 3500 y 3300 cm-1. Las aminas primarias presentan dos bandas (simétrica y asimétrica), las secundarias una sóla banda.
Flexión N-H: Aminas primarias dos bandas a 1640 y 1560 cm-1. Secundarias una banda a 1500 cm-1
Espectro IR de la 1-hexano-amina
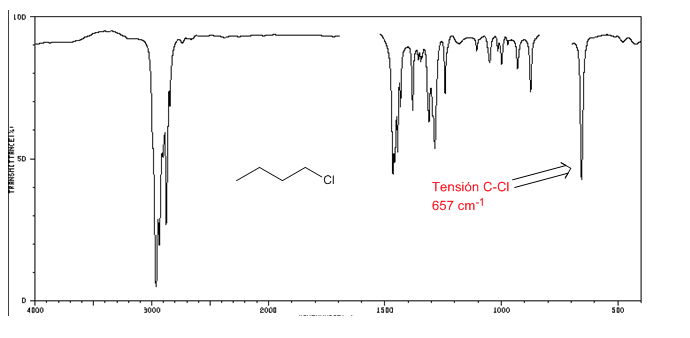
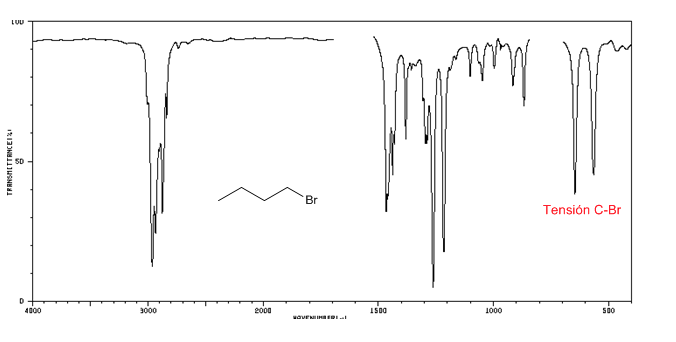
Espectro IR de halogenuros de alquilo
Tensión C-Cl: 785 - 540 cm-1
ensión C-Br: 650 - 510 cm-1
 Fuentes recomendadas para expandir la busqueda:
Fuentes recomendadas para expandir la busqueda:
1. Aga Fano S.A. Espectroscopia de Emisión. (acceso 10 de septiembre del 2007).http://hiq.aga.com.co/International/Web/LG/CO/likelgspgco.nsf/DocByAlias/anal_icp.
2. Alonso, P. et al. quimicaCou..Ed. Mc Graw-Hill. 1990.
3. Álvarez Jiménez, M. D. y Gómez del Río, M. I. Guía didacticaQuímica Analítica II. UNED. 1999.
4. Arribas Jimeno Siro; Burriel Barcelo Fernando; Hernández Méndez Jesús; Lucena Conde Felipe. Química Analítica Cualitativa. ISBN: 8497321405. ISB. 2006.
5. Ayres, Gilbert H. Análisis Químico Cuantitativo. Ediciones del Castillo, 4ta ed. ISBN: 8421902806. 1981.
6. Bermejo Barrera. M del Pilar. Química analítica general, cuantitativa e instrumental. Editorial Paraninfo. 7ma Edición. ISBN: 8428318093. 1990.
7. Blanco, M., Cerdá, V. y Sanz Medel, A., Espectroscopia Atómica Analítica, Publicaciones de la universidadAutónoma de Barcelona. 1990.
8. Brode. R.W, Chemical spectroscopy, Nueva York 1952.
9. Burriel, M.F., Lucena, C.F.Química Analítica Cuantitativa. Edición Revolucionaria. La Habana.1978.
10. Burriel, F. Química Analítica Cualitativa. Editorial Paraninfo. ISBN: 8497321405. pp 1072. , 2003.
- Detalles
- Alejandro Savin.
- Visto: 39360
Estereoquímica y análisis conformacional
La Estereoquímica es la rama de la química que se ocupa de los aspectos tridimensionales de las moléculas y de su reactividad. No se puede entender la Química Orgánica sin tener en cuenta la Estereoquímica. Los sistemas biológicos son muy selectivos y a menudo discriminan moléculas con diferencias estereoquímicas muy pequeñas.
El análisis conformacional forma parte del estudio espacial de las moléculas, esto es, de la Estereoquímica.
CONSTITUCIÓN, CONFIGURACIÓN Y CONFORMACIÓN
Se denominan isómeros a compuestos con la misma fórmula
empírica pero diferente estructura.
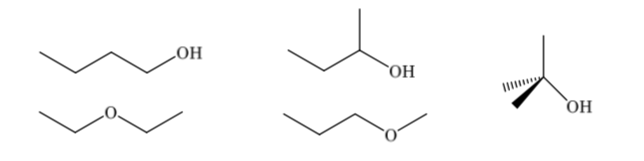
Los isómeros constitucionales difieren en su secuencia de enlace, presentan diferente conectividad. Un compuesto de fórmula molecular C4H10O puede presentar diferentes constituciones:
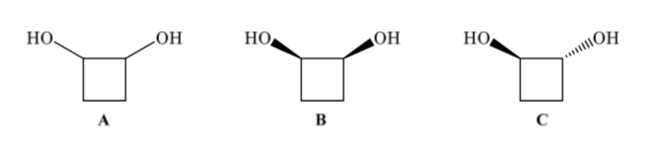
Los isómeros configuracionales tienen la misma conectividad pero difieren en la disposición espacial de los átomos. El 1,2-ciclobutanodiol (A) puede presentar dos isómeros configuracionales según si los dos grupos OH están hacia el mismo lado (B) o lados opuestos (C)
Los Isómeros conformacionales tienen la misma constitución, igual configuración, pero difieren espacialmente en que se pasa de un isómero a otro por simple rotación de un enlace. Las diversas formas que adquieren las moléculas como resultado de la rotación en torno a un enlace sencillo, se llaman conformaciones y cada una de ellas constituye un confórmero.
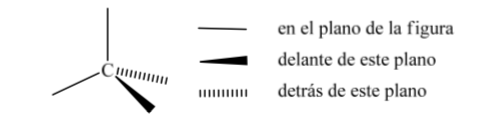
Una de las representaciones más utilizadas del carbono tetraédrico es la llamada representación en perspectiva.
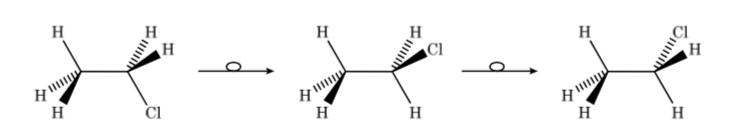
Así, en el caso del cloroetano podemos representar los distintos confórmeros en perspectiva de la forma siguiente:
Los isómeros configuracionales y los conformacionales sonestereoisómeros. Los estereoisómeros tienen la misma conectividad pero difieren en la orientación de alguno de sus átomos en el espacio. Presentan a menudo diferencias considerables en sus propiedades físicas, químicas y biológicas.
CONFORMACIÓN EN MOLÉCULAS ORGÁNICAS ACÍCLICAS
Representación de las moléculas orgánicas
Existen diversas formas de representar en el plano las moléculas.
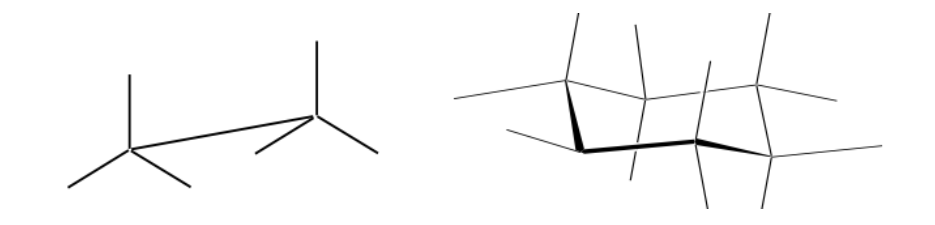
Representación en Perspectiva o Caballete
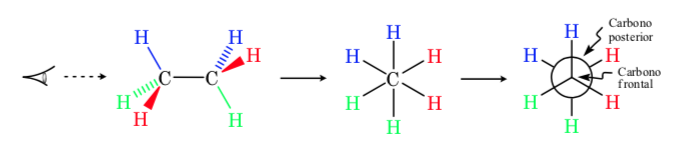
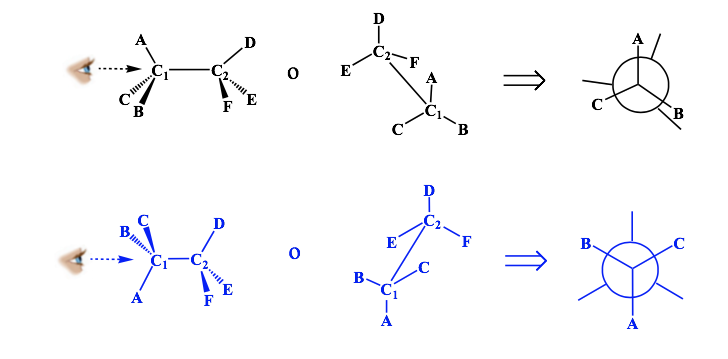
Proyección de Newman
En una proyección de Newman se representan dos átomos de carbono contiguos en una molécula dentro de un círculo. En la parte delantera se sitúa el átomo de carbono que se ve de frente y por detrás del círculo el átomo de carbono solapado. Del centro de círculo salen por delante y por detrás los tres sustituyentes de cada carbono
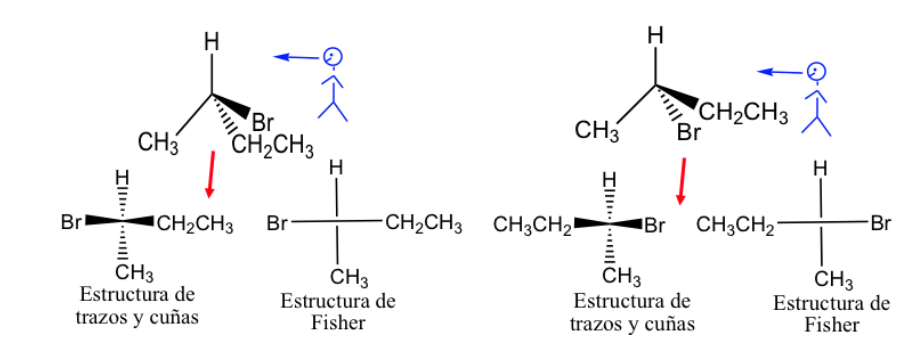
Representación de Fischer
En la representación de Fischer un átomo de carbono tetraédrico se encuentra representado por el centro de una cruz y los sustituyentes se colocan en los extremos de forma que los trazos verticales indican que los enlaces van dirigidos hacia atrás del plano del papel y los horizontales hacia delante. La cadena principal se dispone de forma vertical.
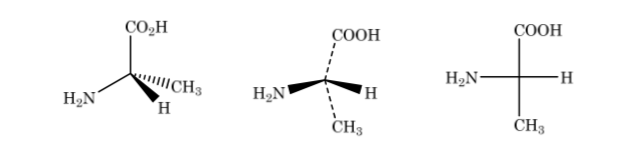
Como se muestra anteriormente la representacion del ácido 2-aminopropanoico
Si el compuesto presenta diferentes átomos de carbono, la cadena carbonada se dibuja en vertical con los sustituyentes de cada carbono en horizontal manteniendo siempre la norma de que los enlaces verticales van hacia detrás del plano y los horizontales hacia delante.
Conformaciones del Etano
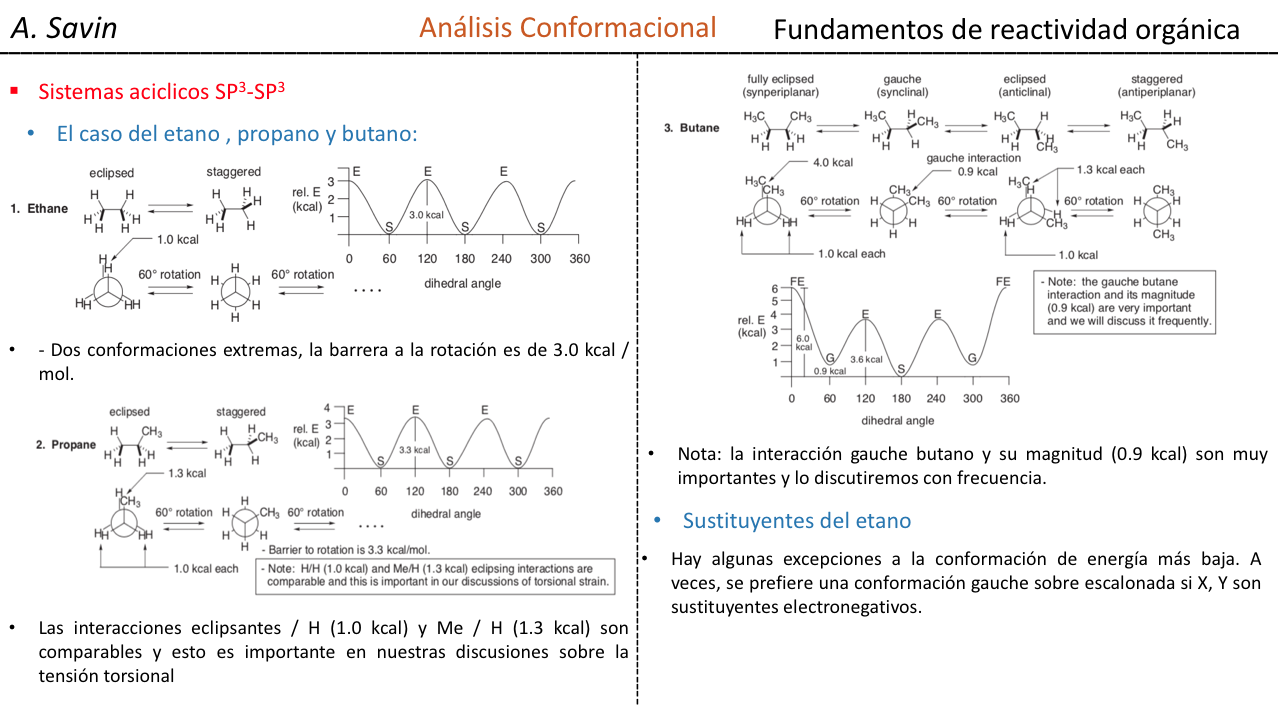
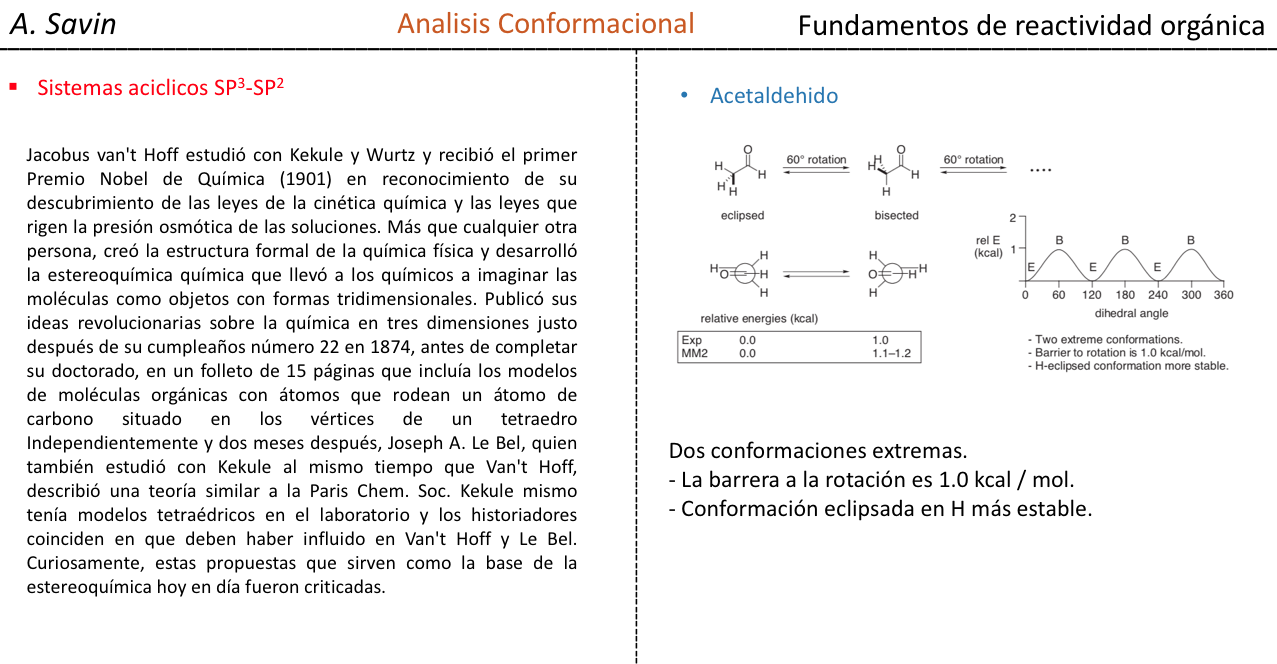
El enlace σ que une los átomos de carbono en el etano es cilíndricamente simétrico y permite la libre rotación alrededor del mismo. La rotación alrededor del enlace C–C interconvierte las diferentes conformaciones del etano. El estudio de su comportamiento termodinámico y cinético se denomina Análisis Conformacional.
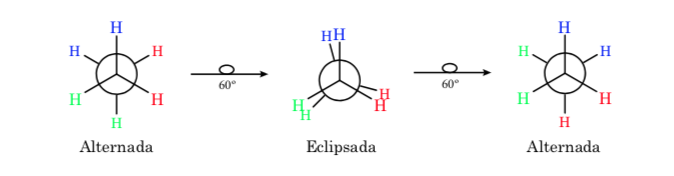
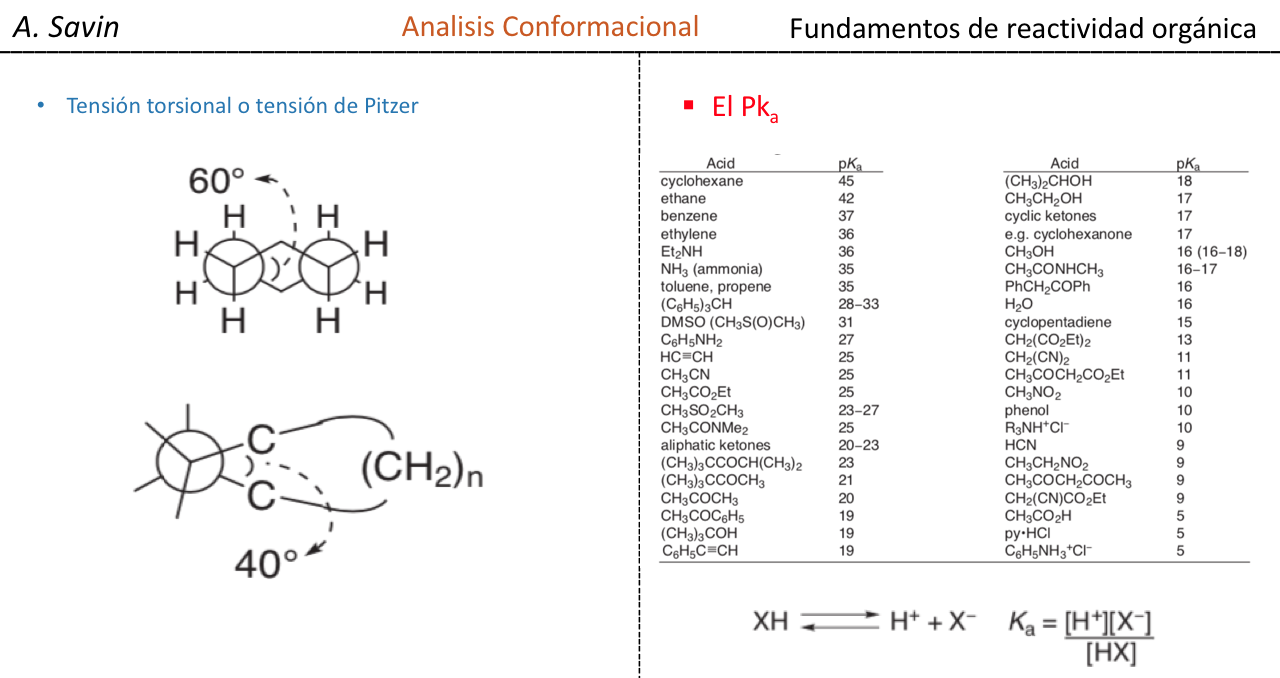
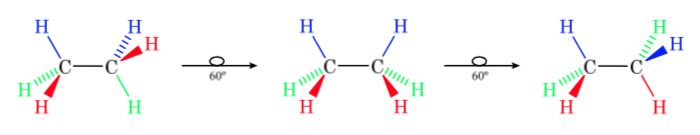
La primera figura representa a la molécula de etano donde los átomos de hidrógeno se encuentran lo más alejados posible con una interacción mínima entre ellos. Se denomina conformación alternada.
La segunda proviene de haber realizado un giro de 60o al enlace C–C y donde los átomos de hidrógeno se encuentran de forma eclipsada.
De nuevo un giro de 60o al enlace C–C nos conduce a una conformación alternada.
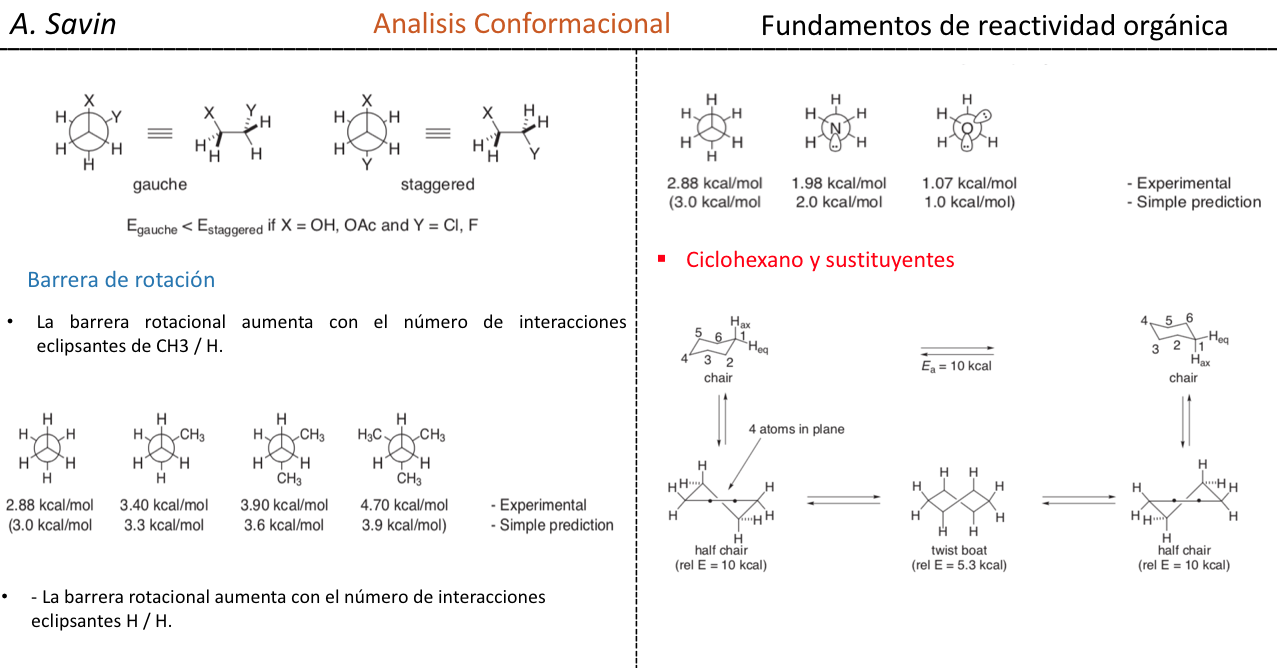
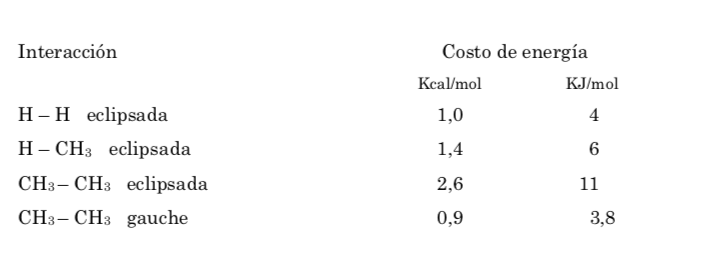
La energía potencial de la molécula es mínima para la conformación alternada, va aumentando con la rotación y alcanza un máximo con la conformación eclipsada. La diferencia energética o barrera de energía a superar para pasar de una conformación a otra es del orden de 3 kcal (ver dibujo).
La mayoría de las moléculas de etano existen naturalmente en la conformación más estable. Como la barrera de energía no es muy alta a temperatura ambiente el número de colisiones con energía suficiente es bastante grande de modo que la interconversión de confórmeros es rápida. La energía requerida para rotar la molécula en torno al enlace C–C se debe a la tensión torsional.
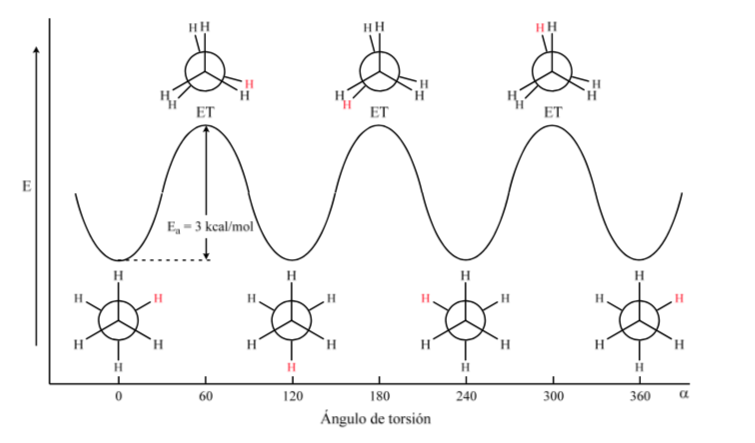
El diagrama anterior representa los cambios de energía al pasar de una conformación a otra durante la rotación del enlace simple C–C.
Cuando se reemplazan los hidrógenos del etano por otros átomos o grupos irán cambiando los niveles energéticos de las conformaciones y su estudio habrá de realizarse para cada caso.
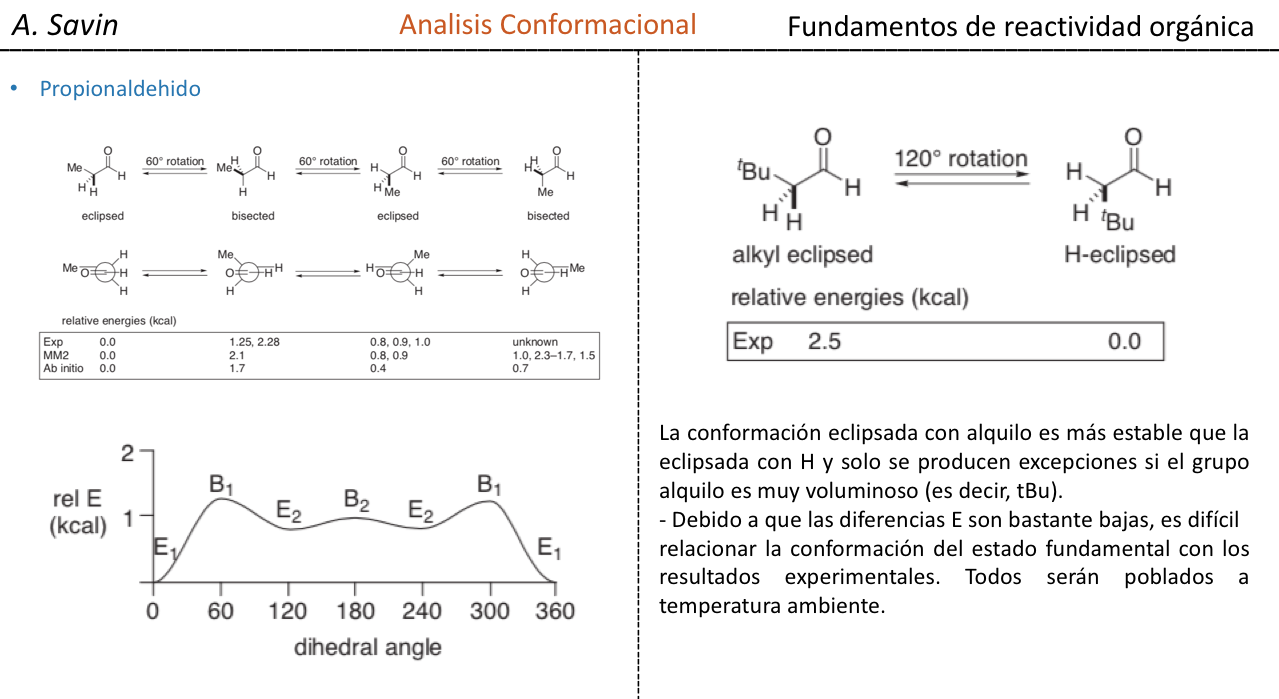
Conformaciones del propano
En el caso de la molécula de propano si representamos las proyecciones de Newman según uno de los enlace C – C y vamos rotando el enlace nos encontramos al igual que en el caso del etano con conformaciones eclipsadas y alternadas.
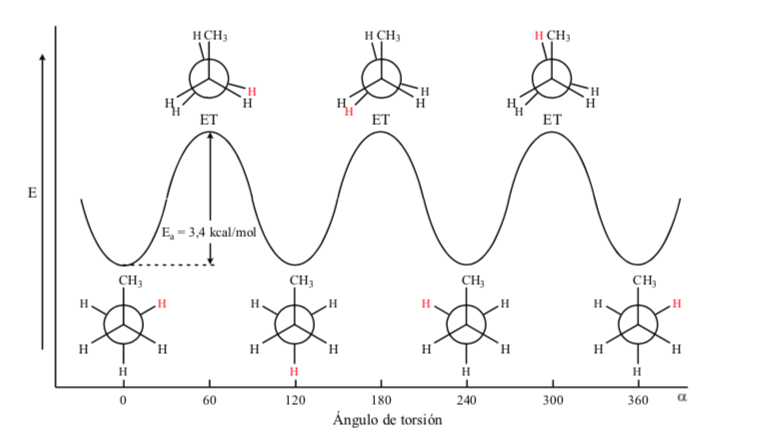
La barrera de energía entre una forma alternada y una eclipsada es mayor que en el caso del etano (3,4 kcal mol-1) ya que el grupo metilo es más grande que el hidrógeno y las interacciones son mayores. Las conformaciones alternadas son más estables que las eclipsadas.
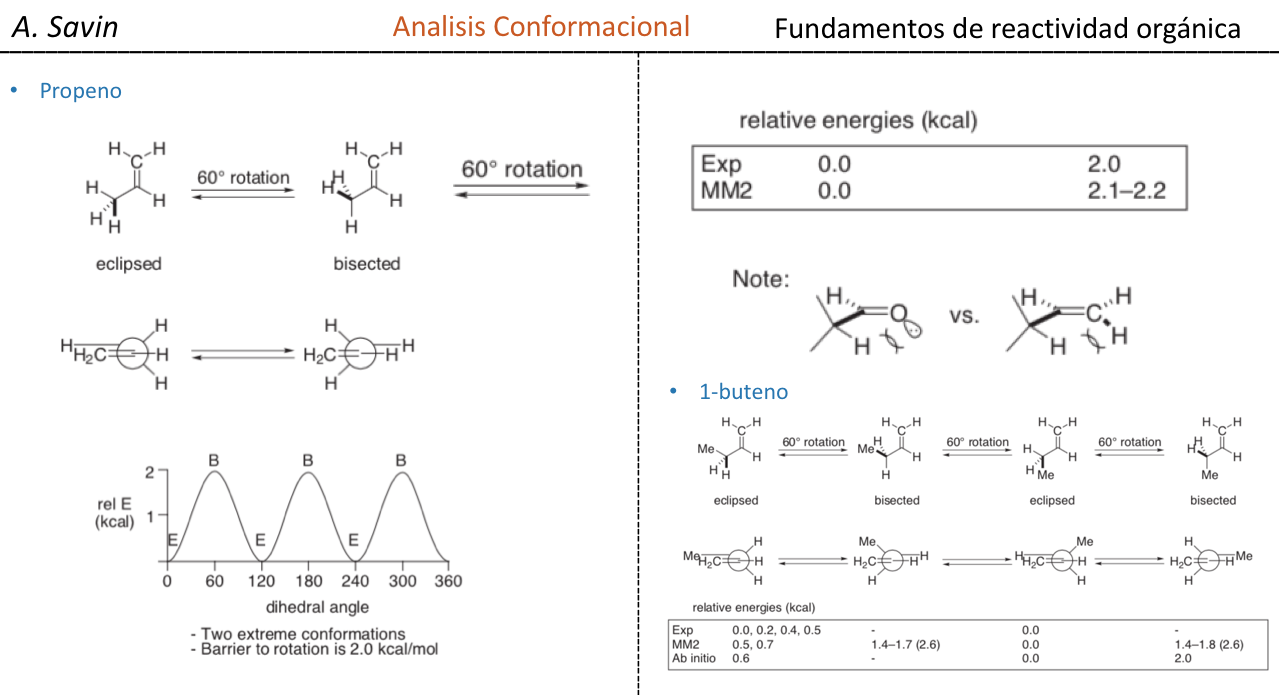
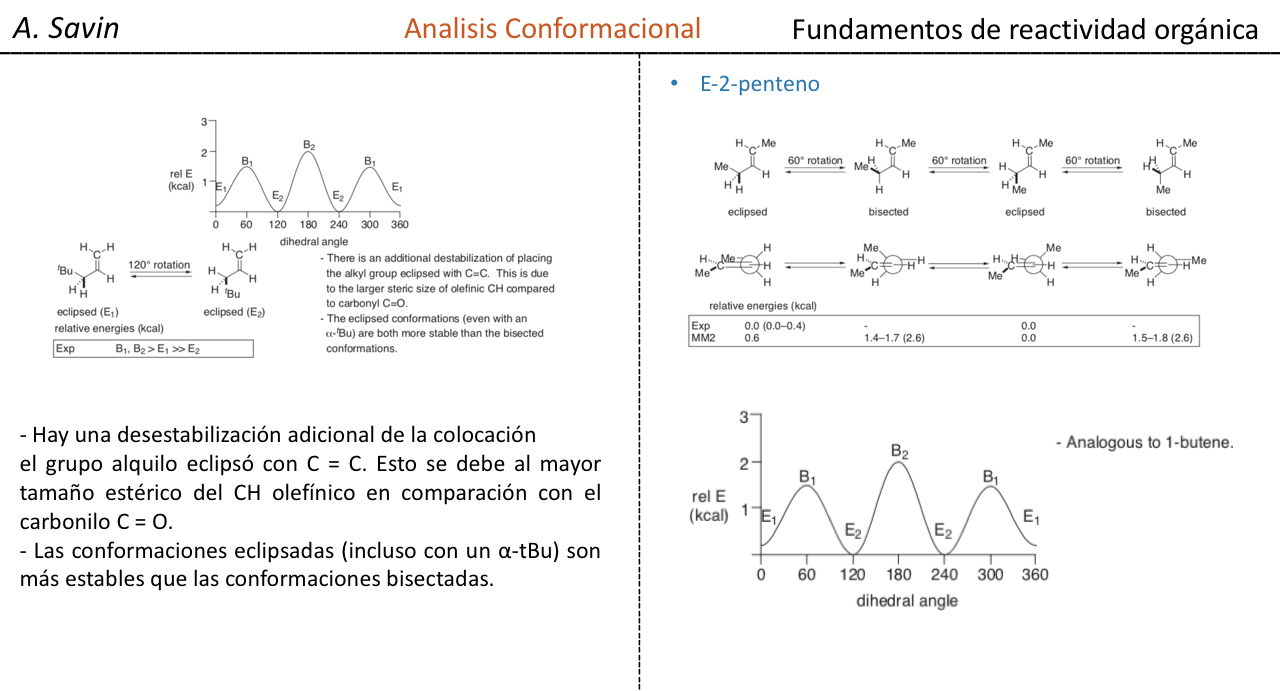
Conformaciones del n-Butano
Si nos fijamos en el enlace central C–C podremos considerar al butano a la hora de representarlo en Newman como una molécula similar a la de etano en la que se han reemplazado dos átomos de hidrógeno por dos grupos metilo.
Al igual que en el etano las conformaciones alternadas tienen menor energía y son más estables que las eclipsadas: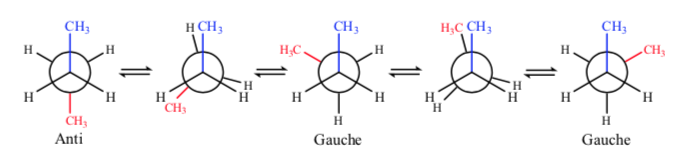
De todas las conformaciones alternadas aquella que tiene los dos grupos metilo en posición lo más opuesto posible (ángulo diedro 180o) se denomina conformación anti y es la más estable. Las otras dos conformaciones alternadas son de energía similar entre ellas y se denominan conformaciones gauche donde los grupos metilo se encuentran formando un ángulo diedro de 60o uno de otro. Existen dos conformaciones de este tipo según la rotación alrededor del enlace C–C.
De las tres conformaciones eclipsadas destacamos en primer lugar aquella donde se encuentran eclipsados los dos grupos metilo que genera mayor interacción y por tanto menor estabilidad. Las otras dos conformaciones eclipsadas presentan menor contenido energético y son algo más estables. Ver el diagrama energético.
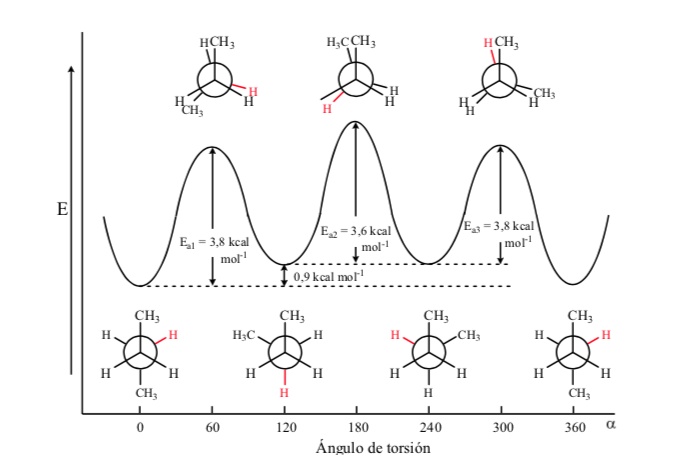
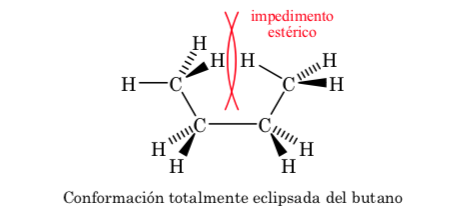
Las colisiones proporcionan la energía necesaria para superar la barrera de la energía de activación.
CONFORMACIÓN EN MOLÉCULAS ORGÁNICAS CÍCLICAS
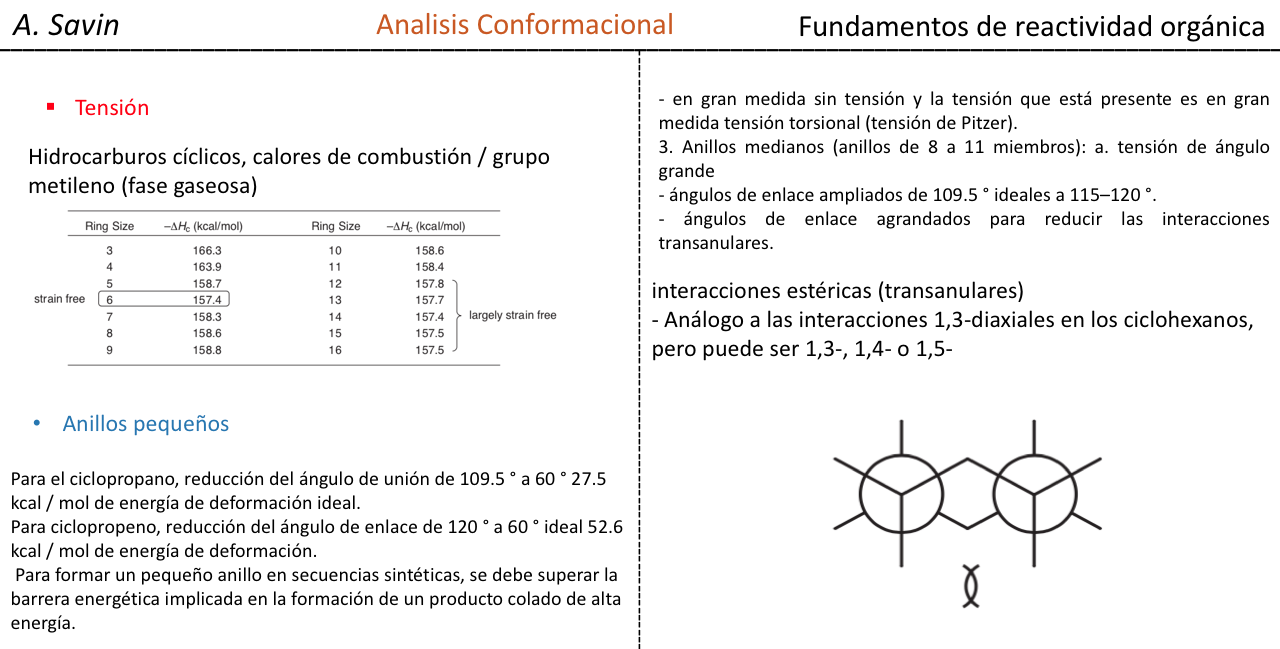
Los cicloalcanos están sometidos a una tensión de anillo llamada Tensión de Bayer debido a la estructura cíclica que presentan. Dicha tensión es el resultado de tres factores:
- Tensión de Enlace
- Eclipsamiento de átomos y enlaces- Tensión Estérica
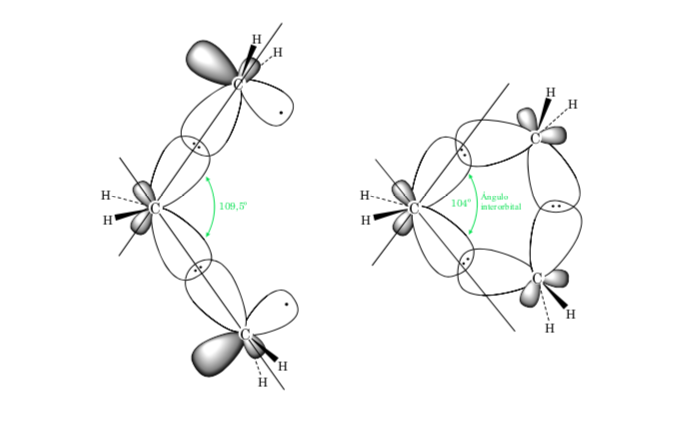
A. La Tensión de Enlace en cicloalcanos es debida a la diferencia en el solapamiento de los orbitales respecto a un alcano acíclico. Cuanto más cercano es el valor del ángulo C–C–C al tetraédrico (109,5o) mayor será el solapamiento y menor la tensión de enlace.
El efecto estérico se debe a la interacción de los átomos a través del espacio. Para evitar que la interacción entre los átomos sea elevada, la molécula adquiere una conformación preferente, esto es la más estable. Para reducir las tensiones y conseguir así estabilidad, la conformación preferente será aquella en la que los grupos voluminosos se alejan lo más posible unos de otros.
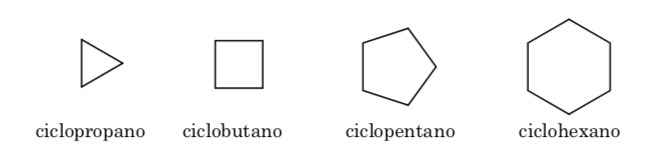
CONFORMACIÓN EN CICLOS DE TRES, CUATRO Y CINCO ÁTOMOS DE CARBONO
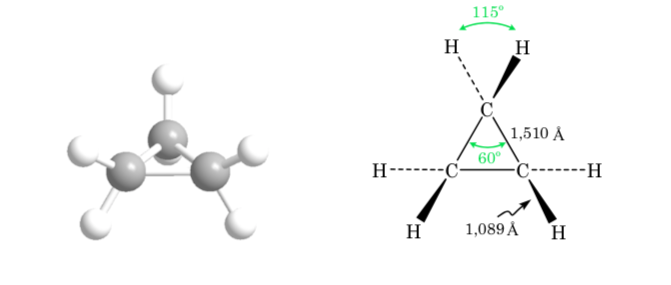
Ciclopropano
La desviación del ángulo de enlace del anillo de tres miembros respecto al ángulo correspondiente a una hibridación sp3 es grande, por lo que el ciclopropano presenta una tensión angular muy alta.
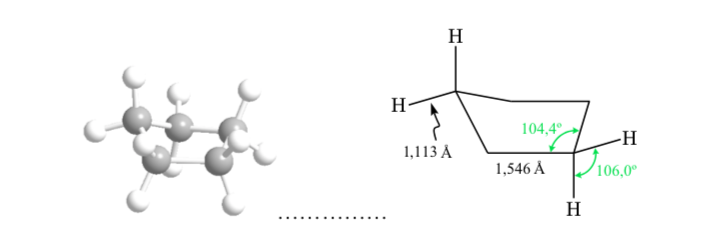
Ciclobutano: Conformación mariposa
La molécula presenta una estructura ligeramente alabeada. Los enlaces C–C están menos curvados y su reactividad es menor que en el caso del ciclopropano. La interconversión entre las distintas conformaciones es rápida.
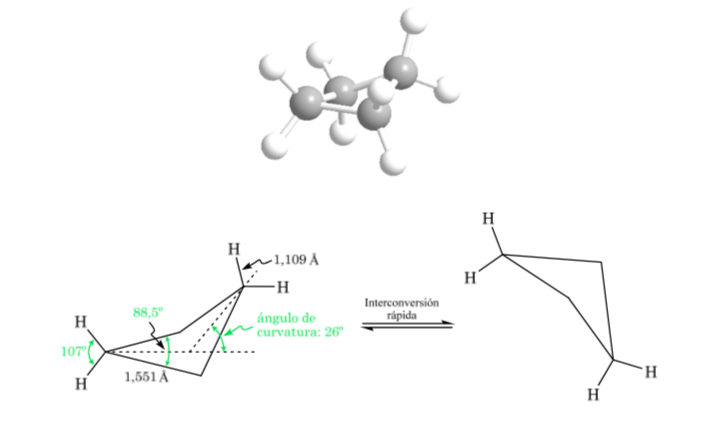
Ciclopentano: Conformación sobre
El ciclopentano si fuera plano presentaría una tensión por eclipsamiento alta, debido a diez interacciones H–H eclipsadas por lo que se produce un plegamiento en el anillo. El plegamiento libera tensión torsional. El ciclopentano no presenta la reactividad de los anillos de tres y cuatro miembros.
ANÁLISIS CONFORMACIONAL DE CICLOHEXANOS : Conformación silla
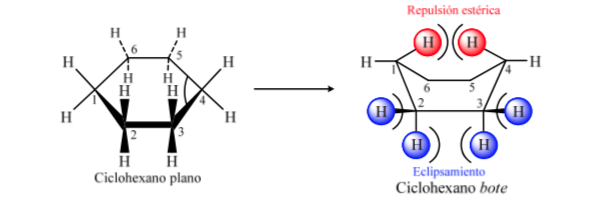
Si el ciclohexano presentara una estructura plana le correspondería un ángulo de enlace de 120o. Un carbono con hibridación sp3 y sin tensión presenta un ángulo tetraédrico de 109,5o, lo que hace que el ciclohexano adquiera una conformación no plana y más estable: Conformación silla.
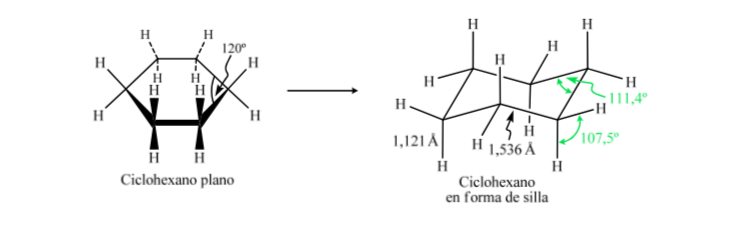
El ciclohexano en conformación silla presenta 6 átomos de hidrógeno axiales y 6 hidrógenos ecuatoriales. Los hidrógenos axiales están dirigidos hacia arriba y hacia abajo del anillo, mientras que los hidrógenos ecuatoriales se dirigen hacia fuera del anillo.
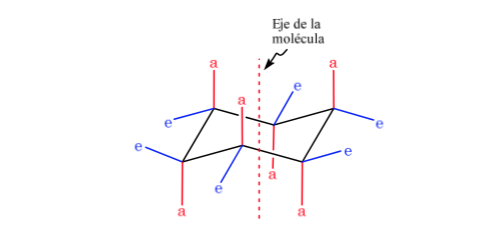
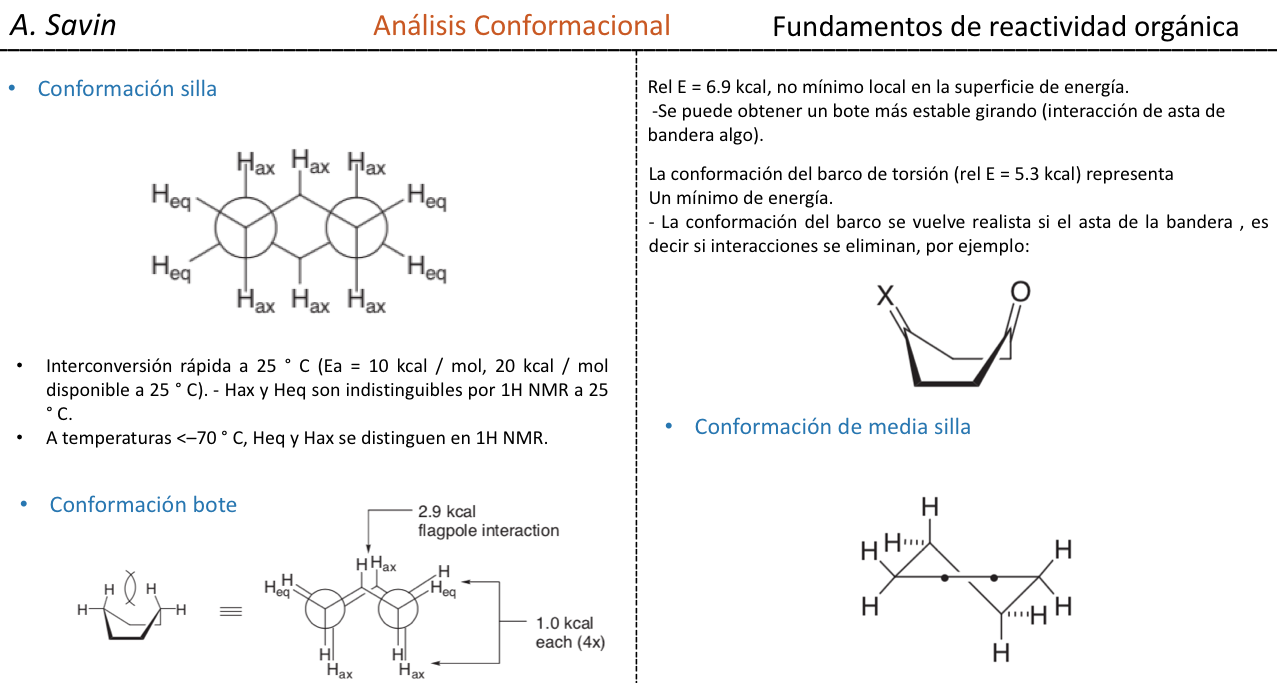
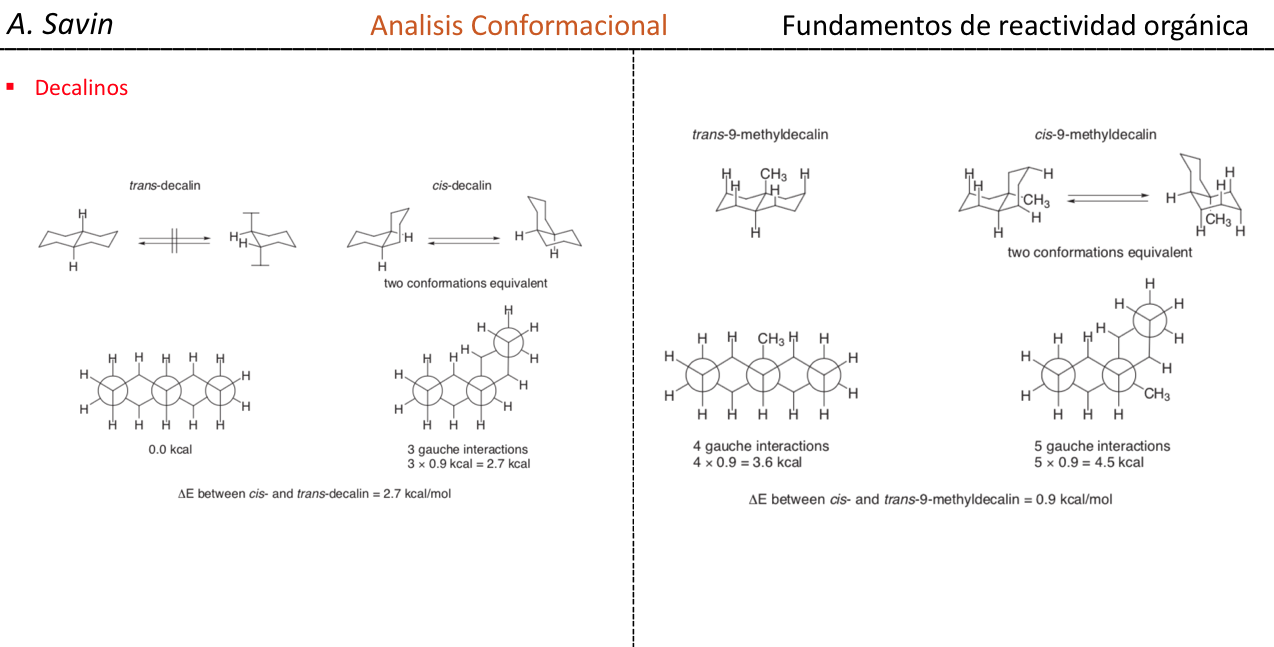
El ciclohexano en conformación silla es susceptible de modificar su conformación por otra silla de igual estabilidad. A este fenómeno se le denomina interconversión de dos formas silla.
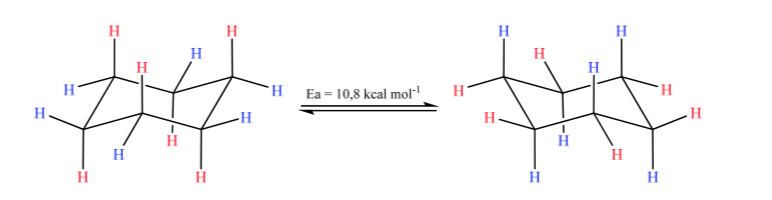
Fijándose detenidamente en la interconversión se observa que el enlace C–H axial pasa a ser ecuatorial y viceversa, el ecuatorial pasa a ser axial.
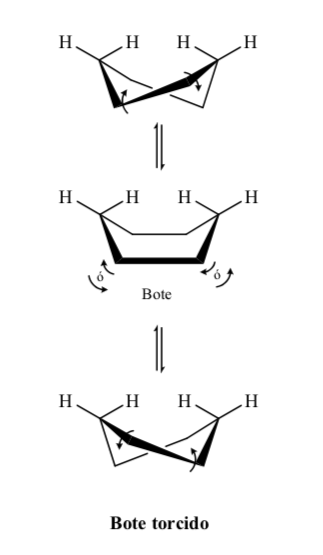
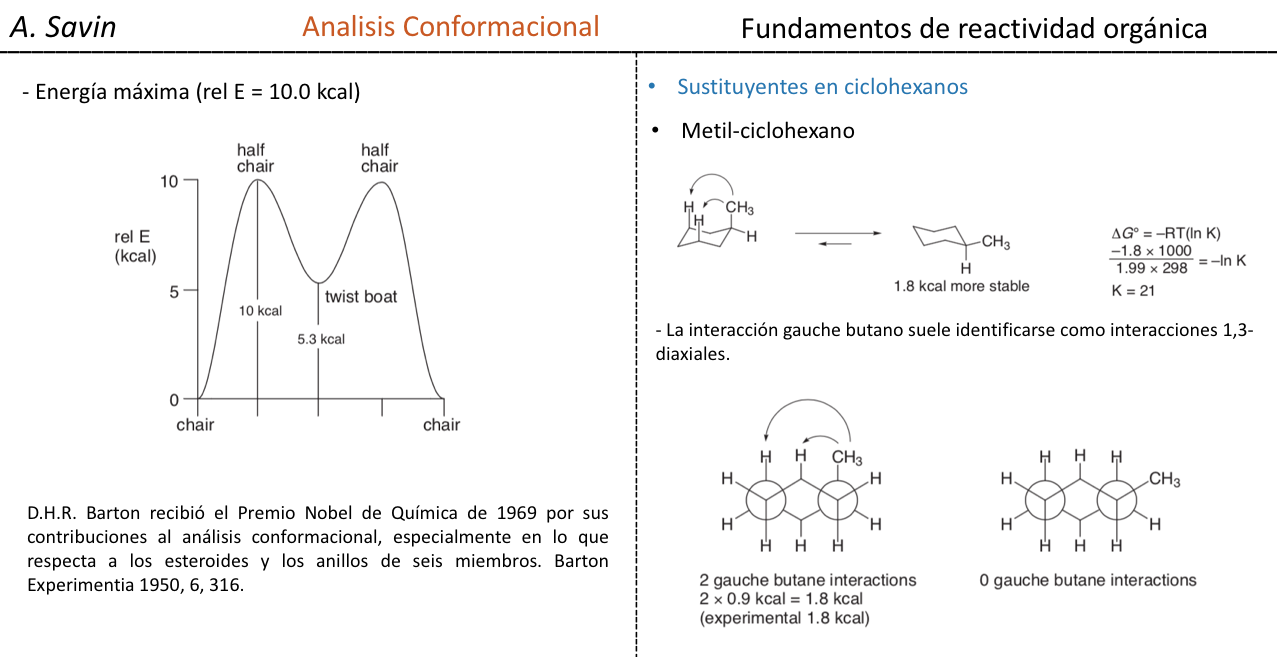
Niveles energéticos de los confórmeros del ciclohexano:
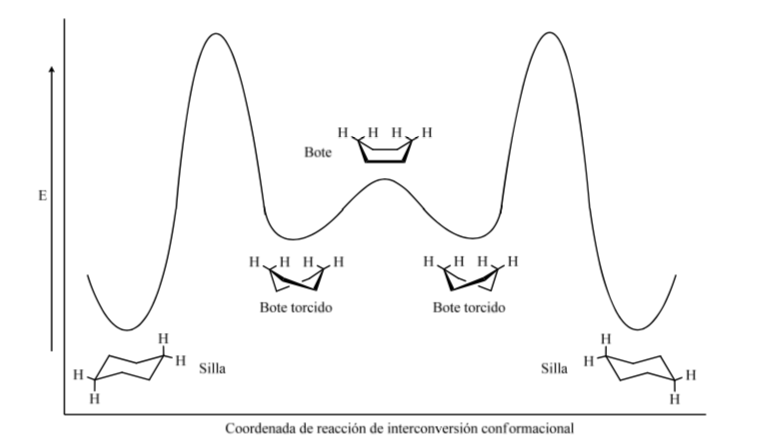
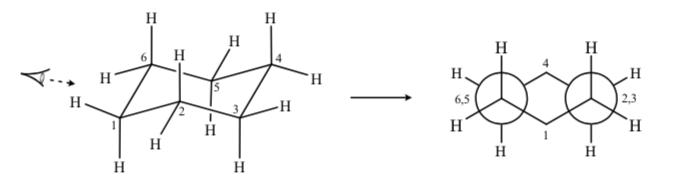
Si observamos una conformación silla del ciclohexano de forma que el átomo C-2 solapa al C-3 y el átomo C-6 solapa al C-5 podemos dibujar una proyección de Newman del ciclohexano:
CICLOHEXANOS SUSTITUIDOS
Si consideramos el metilciclohexano podemos proponer dos estructuras que lo representen según si el grupo metilo se encuentra en posición axial o ecuatorial.
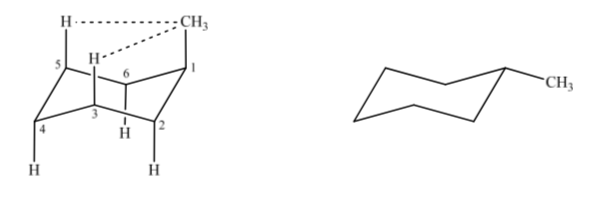
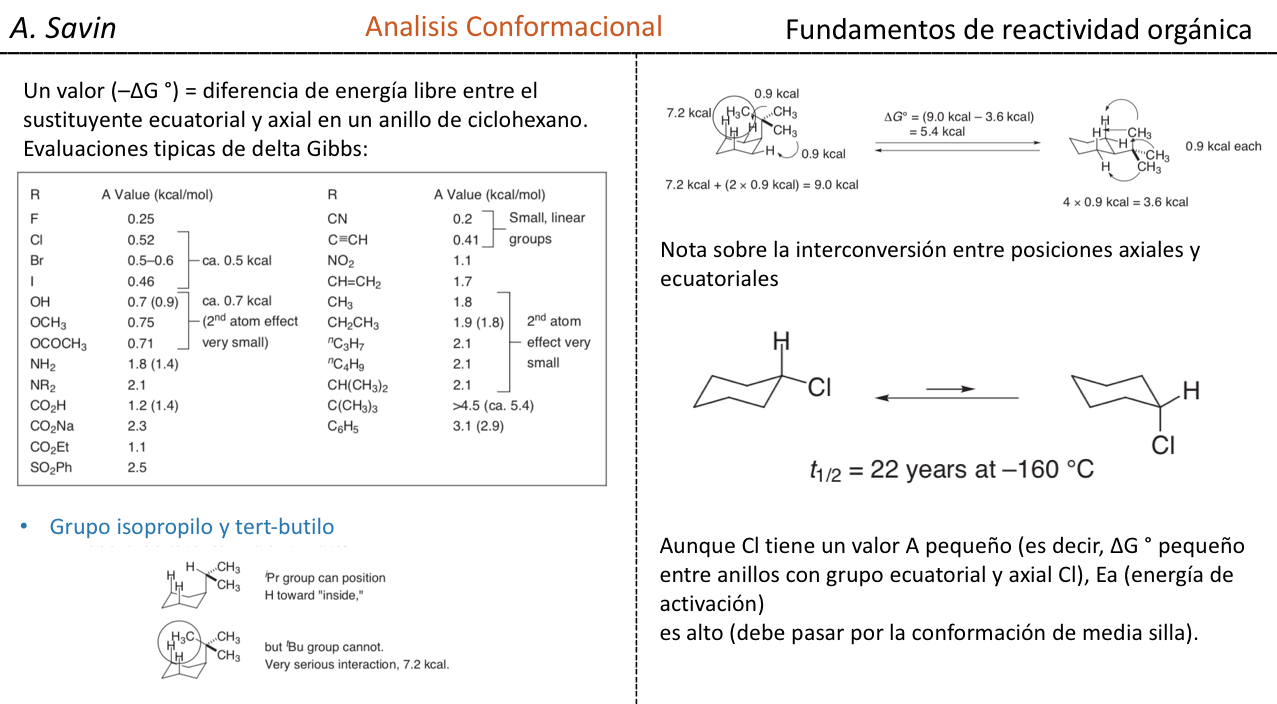
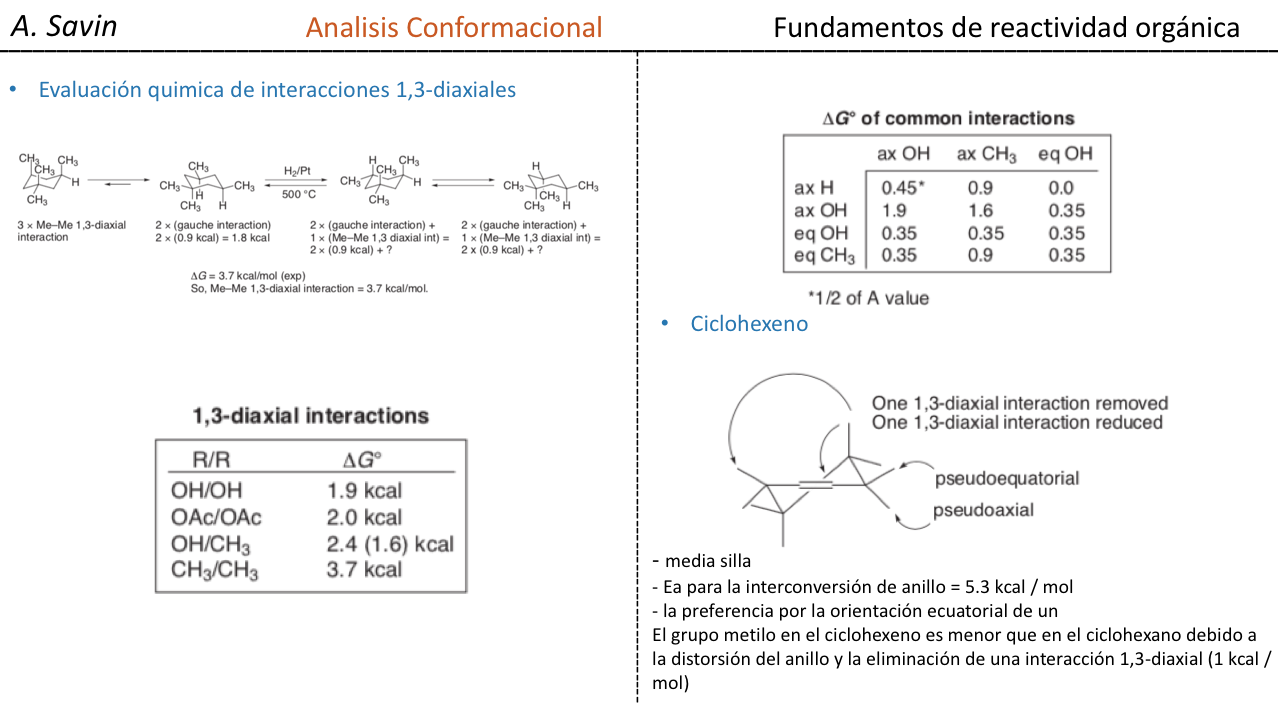
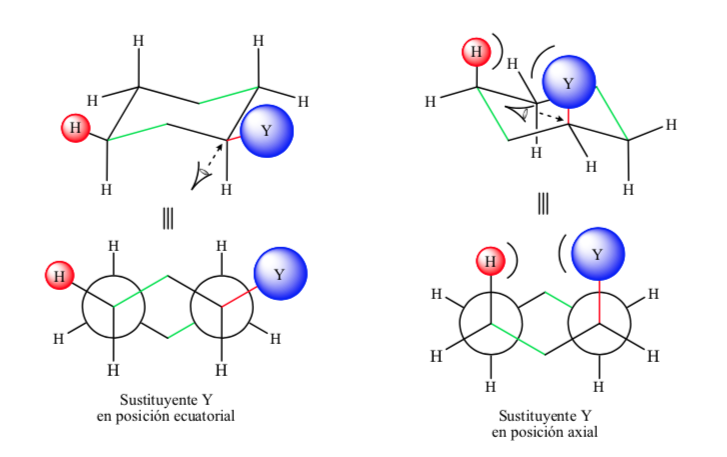
Si el sustituyente diferente al hidrógeno se encuentra en posición axial se producen interacciones con los hidrógenos en posición 3 y 5 del anillo por proximidad espacial. Si se encuentra en posición ecuatorial el sustituyente va dirigido hacia fuera del anillo y las interacciones son menores. Tal como era de esperar se encuentra que la conformación con el sustituyente en ecuatorial es alrededor de 1,8 kcal más estable que si el sustituyente se encuentra en posición axial. Así se dice que un sustituyente en posición axial presenta una interacción 1,3-diaxial que lo hace menos estable que en posición ecuatorial.
En la siguiente figura se representan el Newman las dos conformaciones de un ciclohexano sustituido tanto con el sustituyente en posición ecuatorial como en axial.
Ciclohexanos disustituidos
Si tenemos un ciclohexano dimetilsustituido podemos encontrarnos con diferentes posibilidades:
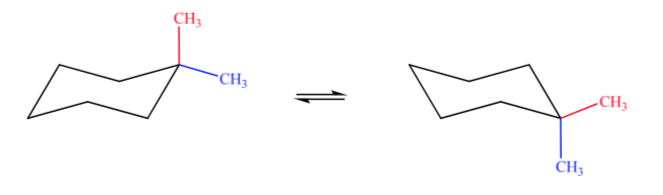
A. 1,1-dimetilciclohexano B. 1,2-dimetilciclohexano C. 1,3-dimetilciclohexano D. 1,4-dimetilciclohexano
En el caso A un grupo metilo se encontrará en posición axial y el otro en ecuatorial por lo que las dos conformaciones posibles serán igual de estables.
En cada uno de los casos restantes podemos representar dos estructuras diferentes según si los dos metilos se encuentran en cis (dirigidos hacia el mismo lado del plano) o en trans (dirigidos hacia lados opuestos).
En B el isómero cis debe tener los dos metilos hacia el mismo lado del plano central por lo que un metilo será axial y el otro ecuatorial.
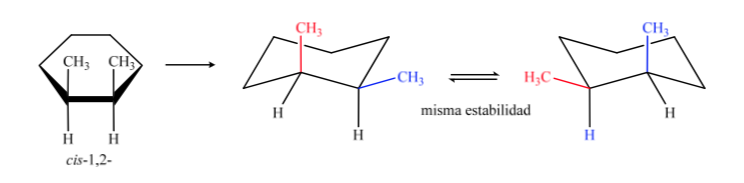
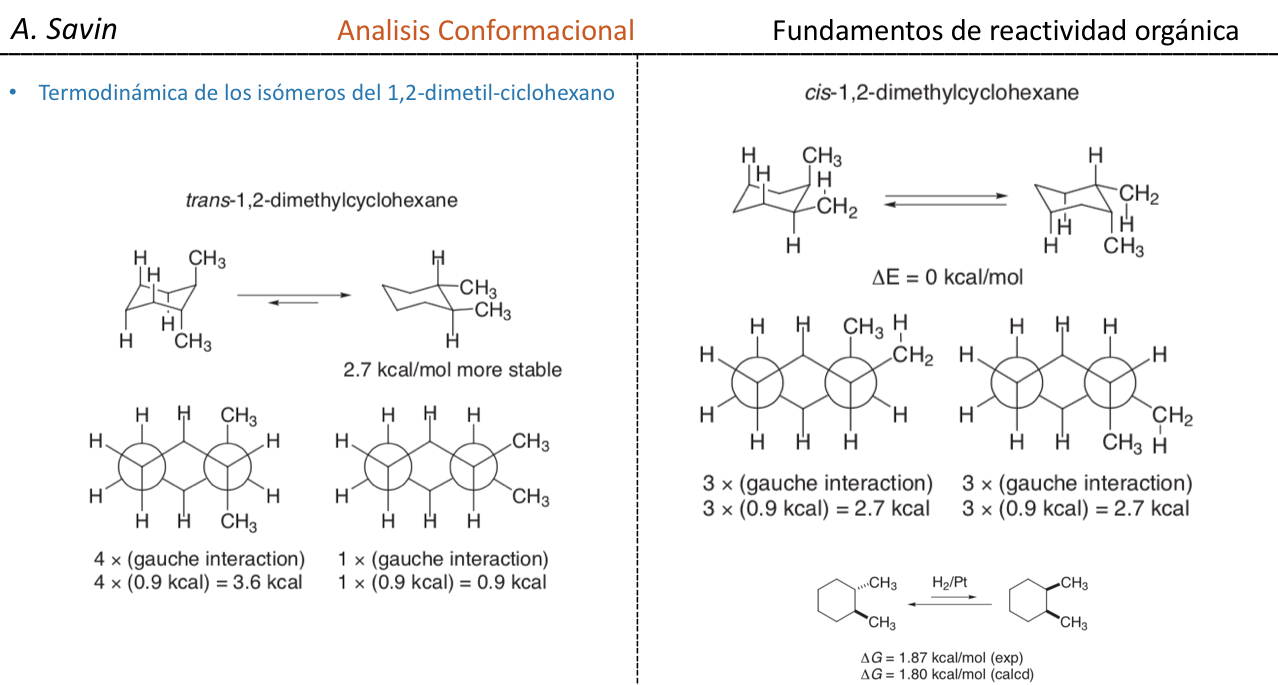
El isómero trans es el que tiene los dos metilos hacia lados opuestos por lo que presentará los dos metilos en posición axial o los dos en ecuatorial. La conformación que presente los dos en ecuatorial es mas estable que en axial por lo tanto podremos decir que el isómero trans-1,2-dimetilciclohexano es más estable que el isómero cis-1,2 –dimetilciclohexano.
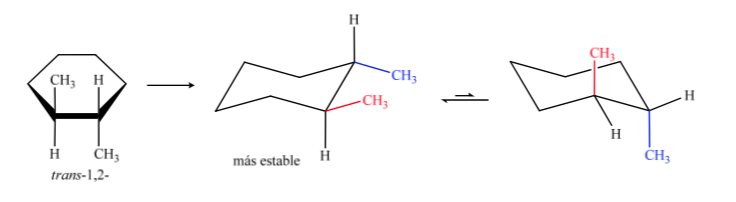
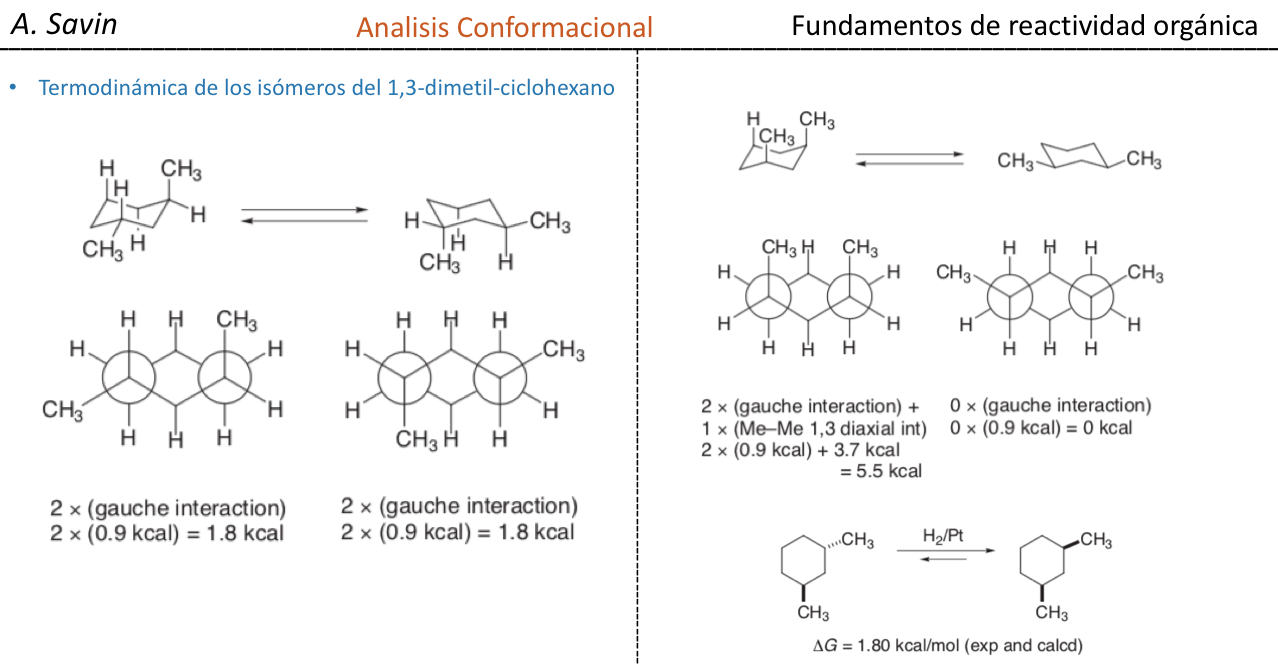
En el caso del 1,3-dimetilciclohexano podemos representar:
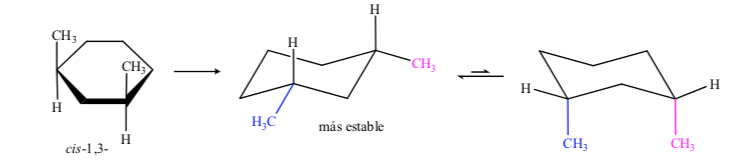
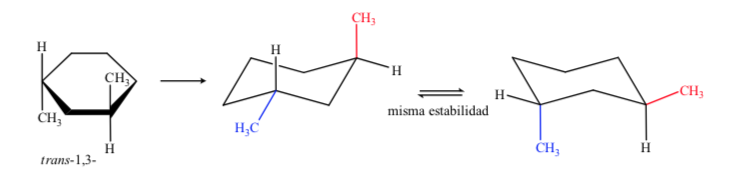
Se concluye que el isómero cis, que presenta los dos sustituyentes en ecuatorial, es más estable que el trans, que siempre tendrá un metilo en axial y otro en ecuatorial.
En el caso D, 1,4-dimetilciclohexano tendremos:
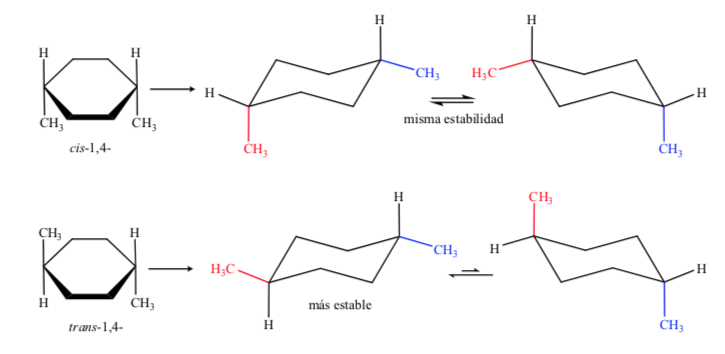
En este caso el isómero trans es más estable por disponer los dos metilos en ecuatorial.
Cuando se trata de ciclohexanos disustituidos con grupos diferentes el isómero más estable será el que tenga el grupo más voluminoso en ecuatorial
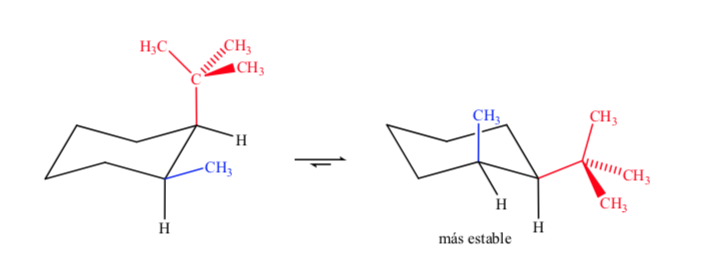
Finalmente, en el caso de ciclohexanos polisustituidos que presenten sustituyentes diferentes la conformación preferente será la que tenga el mayor número de grupos voluminosos en posición ecuatorial.
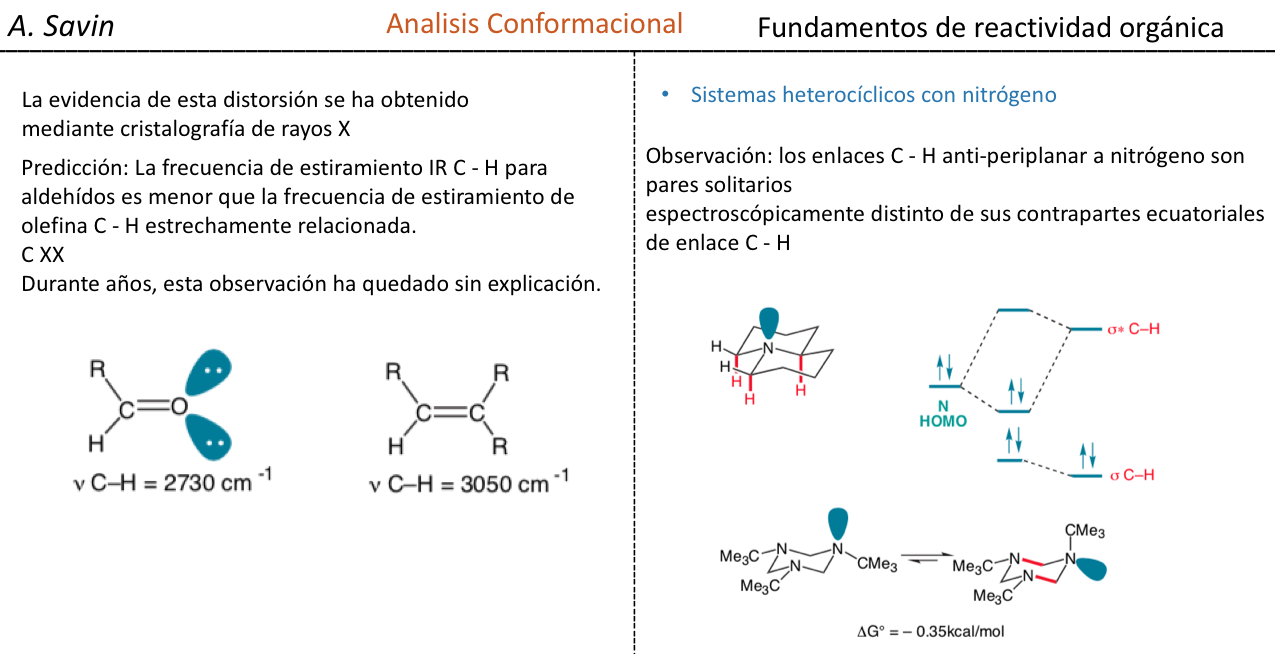
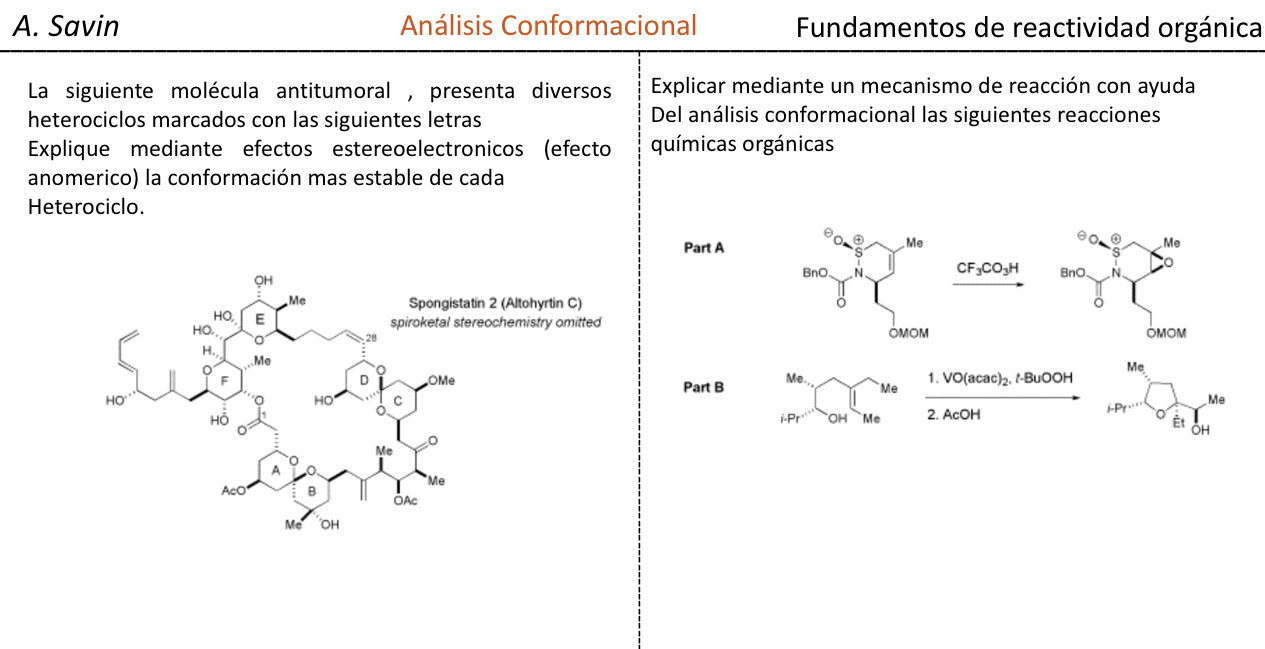
CONFORMACIÓN EN HETEROCICLOS DE SEIS MIEMBROS:
EFECTO ANOMÉRICO
En química orgánica, el efecto anomérico o efecto Edward-Lemieux es un efecto estereoelectrónico que describe la tendencia de los sustituyentes heteroatómicos adyacentes a un heteroátomo en un anillo de ciclohexano a preferir la orientación axial en vez de la orientación ecuatorial menos cubierta, que se esperaría a partir de consideraciones estéricas. Este efecto fue observado originalmente en anillos de piranosa por J. T. Edward en 1955; en aquel tiempo, N.-J. Chii y Raymond U. Lemieuxcomenzaron a estudiar el equilibrio de anomerización de los derivados totalmente acetilados de algunas aldohexopiranosas. El término "efecto anomérico" fue introducido en 1958.
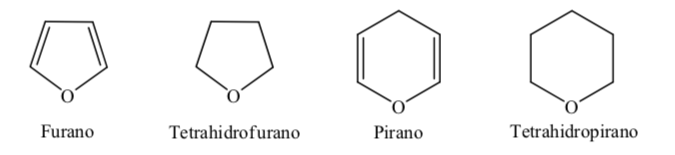
Algunos de los heterociclos de cinco y seis miembros más comunes en los procesos biológicos son:
A. Heterociclos oxigenados:
B. Heterociclos nitrogenados:
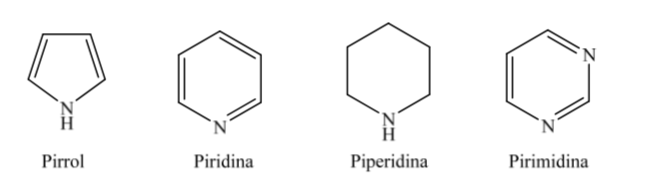
Cuando se trata de heterociclos totalmente hidrogenados de cinco y seis miembros como el tetrahidrofurano y el tetrahidropirano, la conformación más estable que presentan es similar a la del ciclopentano y ciclohexano respectivamente, es decir, conformación sobre o conformaciónsilla.
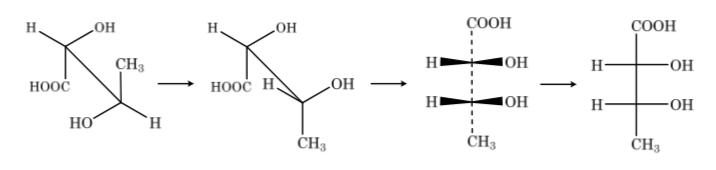
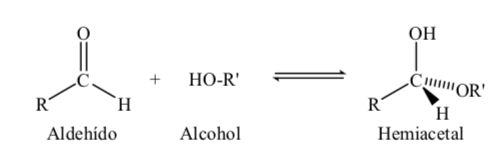
Los azúcares de 5 y 6 átomos de carbono adquieren una estructura cíclica cuando se forma una unión hemiacetálica por reacción del carbonilo (aldehído o cetona del azúcar) con un grupo hidroxilo.
La reacción de formación del hemiacetal podemos esquematizarla de la forma:
Para su estudio, los azúcares se representan mediante estructuras lineales siguiendo las normas de las proyecciones de Fischer.
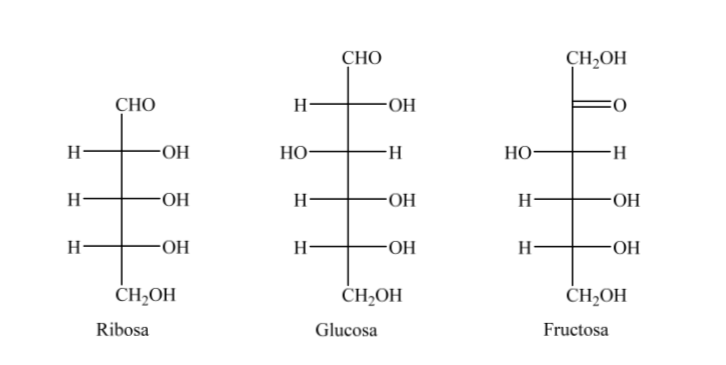
Así, a modo de ejemplo, la ribosa, la glucosa y la fructosa las podemos representar:
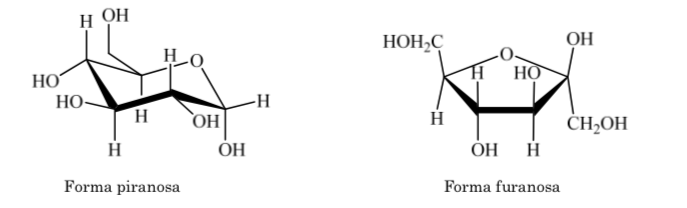
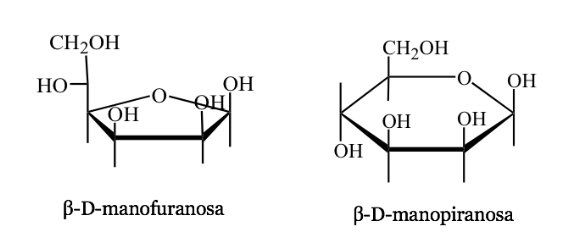
Al formarse el hemiacetal la molécula adquiere una disposición curvada en la que la función aldehído de la aldosa en el carbono 1 se ubica próxima al hidroxilo del carbono 5, produciéndose la adición al doble enlace del carbonilo generando el hemiacetal cíclico de seis miembros (anillo piranósido).
Lo mismo ocurre con las cetosas, pero en este caso la unión hemiacetálica se da entre el carbono 2 y el carbono 5 formándose un anillo de cinco miembros. (anillo furanósido).
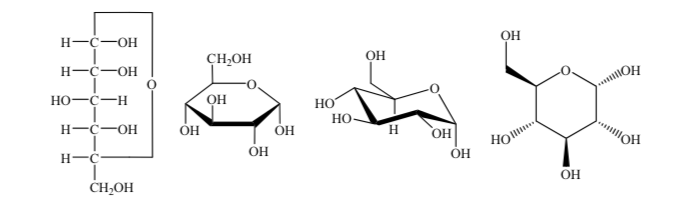
Se utilizan diferentes formas de representar la estructura de la glucosa en forma cíclica:
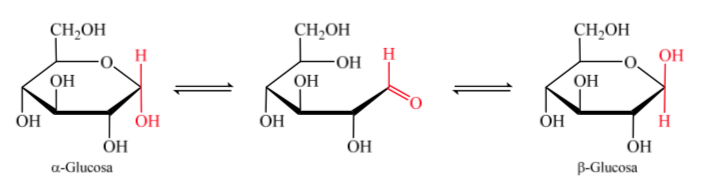
Anómeros
Se denominan anómeros a los isómeros de los monosacáridos de más de 5 átomos de carbono que han desarrollado una unión hemiacetálica lo que les ha permitido tomar una estructura cíclica y determinar 2 diferentes posiciones para el grupo hidroxilo (α o β) en el nuevo centro creado.
El anillo de piranosa debe ser similar al del ciclohexano y existir en conformación silla en preferencia a la de bote torcido para reducir al mínimo las tensiones torsionales. El análisis de rayos X demuestra que estas suposiciones son correctas.
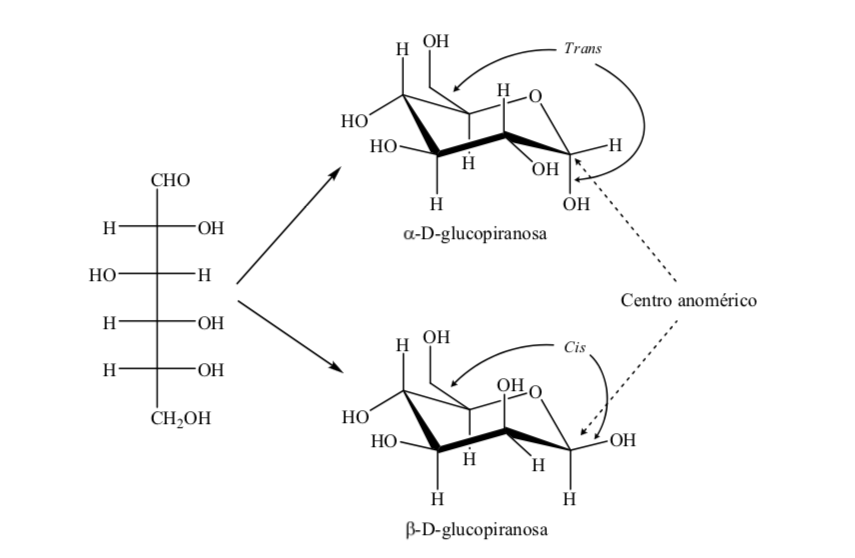
Los dos anómeros son designados anómeros alfa (α) o beta (β), de acuerdo con la relación configuracional entre el centro anomérico y el átomo de referencia anomérico. El centro anomérico está en el carbono hemiacetálico y es el carbono anomérico C-1, que se une a través del oxigeno al C-5, que se une al oxígeno hemicetálico.
Hay sin embargo dos posibles conformaciones silla para un mismo anómero de la D-(+)-glucopiranosa.
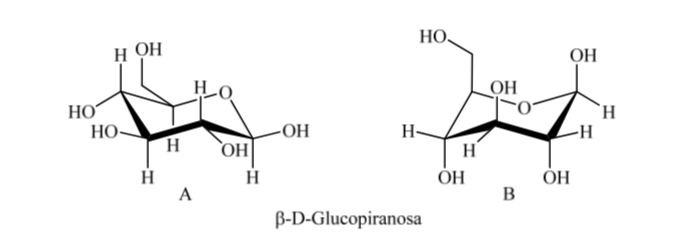
Así, Para la β-D-(+)-glucopiranosa hay dos posibles conformaciones. Será más estable aquella que presenta los grupos más voluminosos en ecuatorial (A).
¿Qué sucede para la α-D-(+)-glucosa? En este caso la conformación más estable es aquella en la que el OH en el carbono anomérico está en axial y los grupos voluminosos en ecuatorial (A).
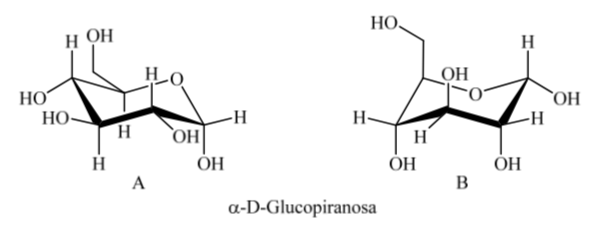
Es interesante observar que de todas las D-aldohexosas, solo la β-D- (+)-glucopiranosa puede asumir una conformación en la que cada uno de sus grupos grandes puede ocupar una posición ecuatorial. Esto concuerda con el hecho de que la β -D-(+)-glucopiranosa sea el azúcar de mayor presencia en la naturaleza.
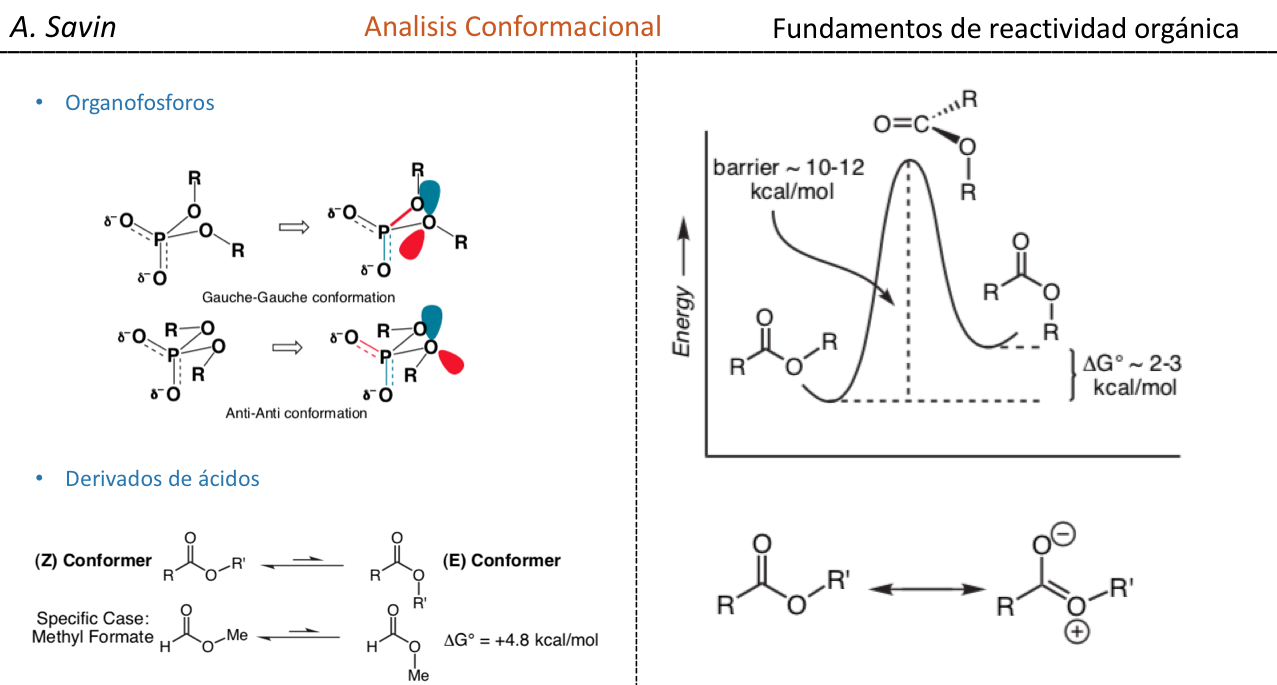
Efectos electronicos
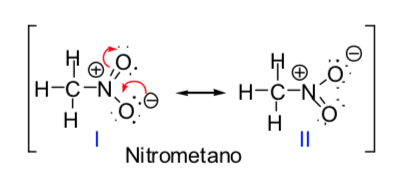
Algunas moléculas orgánicas se pueden representar mediante dos o más estructuras de Lewis, que difieren entre sí únicamente en la colocación de los electrones y que se denominan estructuras resonante s. En estos casos, la molécula tendrá características de ambas estructuras y se dice que en realidad la molécula es un híbrido de resonancia de las estructuras resonantes. El método de la resonancia permite saber, de forma cualitativa, la estabilización que puede conseguir una molécula por deslocalización electrónica. Cuanto mayor sea el número de estructuras resonantes mediante las que se puede describir una especie química mayor será su estabilidad.
El concepto de estructuras resonantes se puede aplicar en la descripción del nitrometano, que se puede representar mediante las dos estructuras de Lewis que se indican a continuación:
Enlaces conjugados y su energia
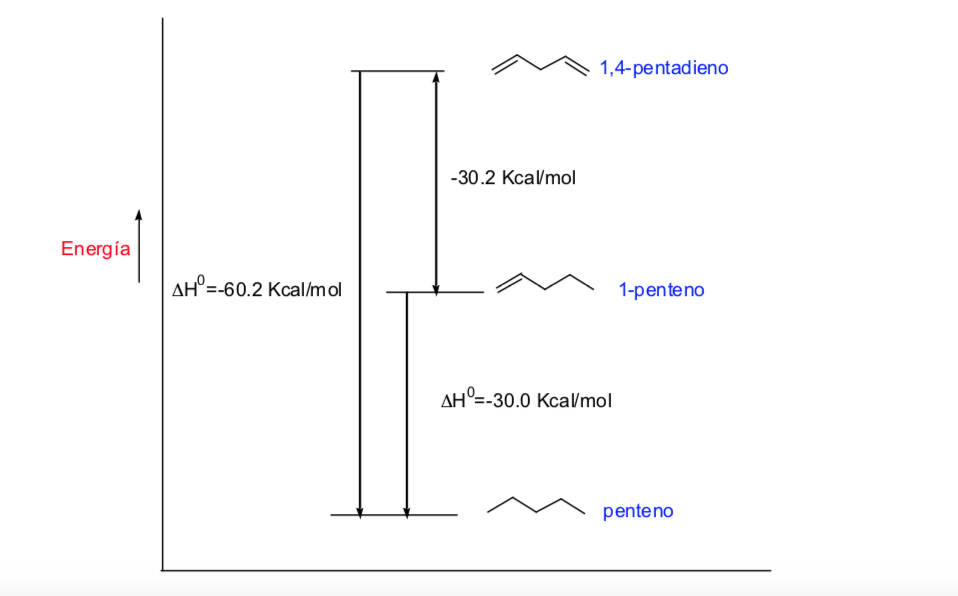
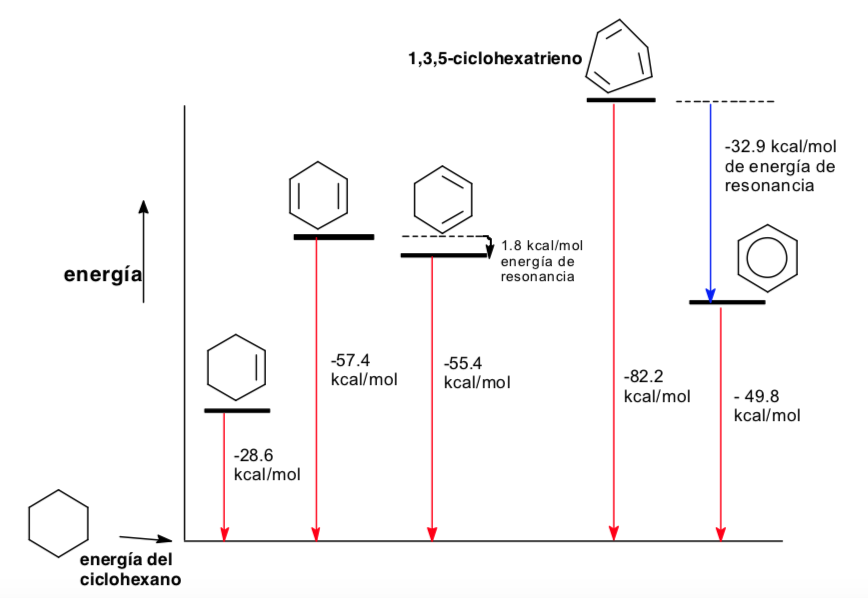
Analisis conformacional de la aromaticidad
 Termodinamica y el analisis conformacional (fectos estereoelectronicos)
Termodinamica y el analisis conformacional (fectos estereoelectronicos)
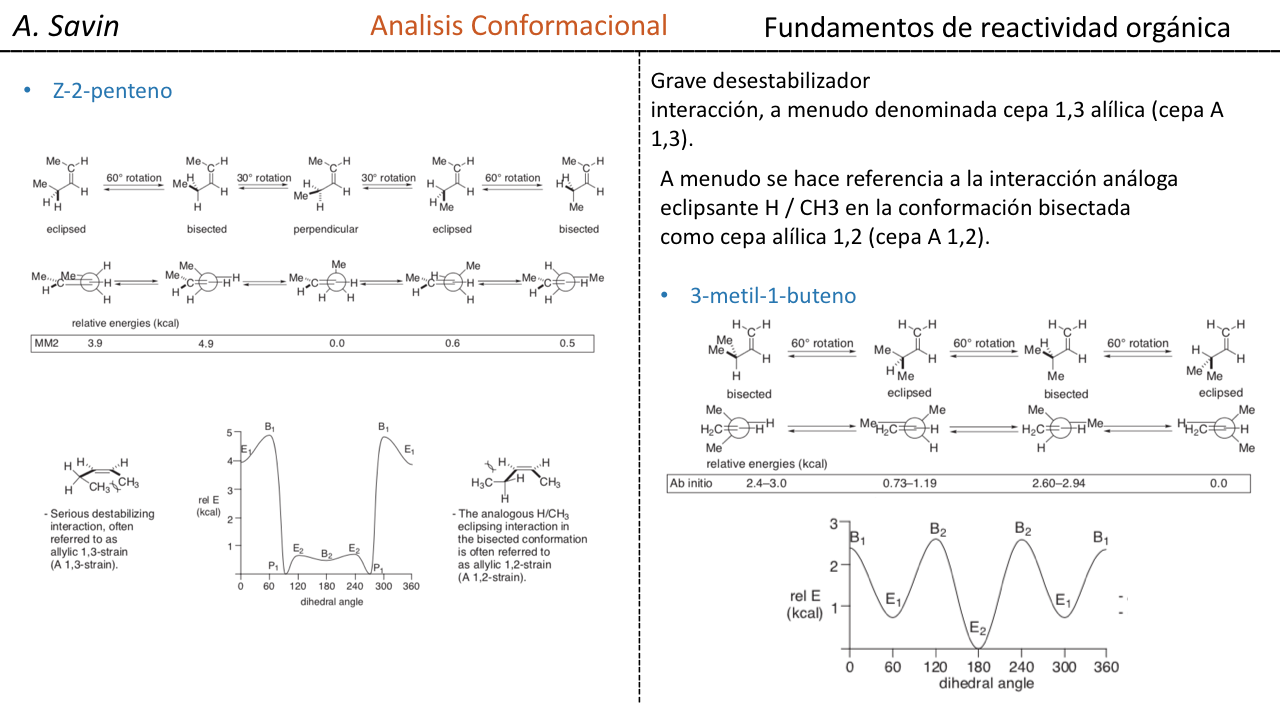
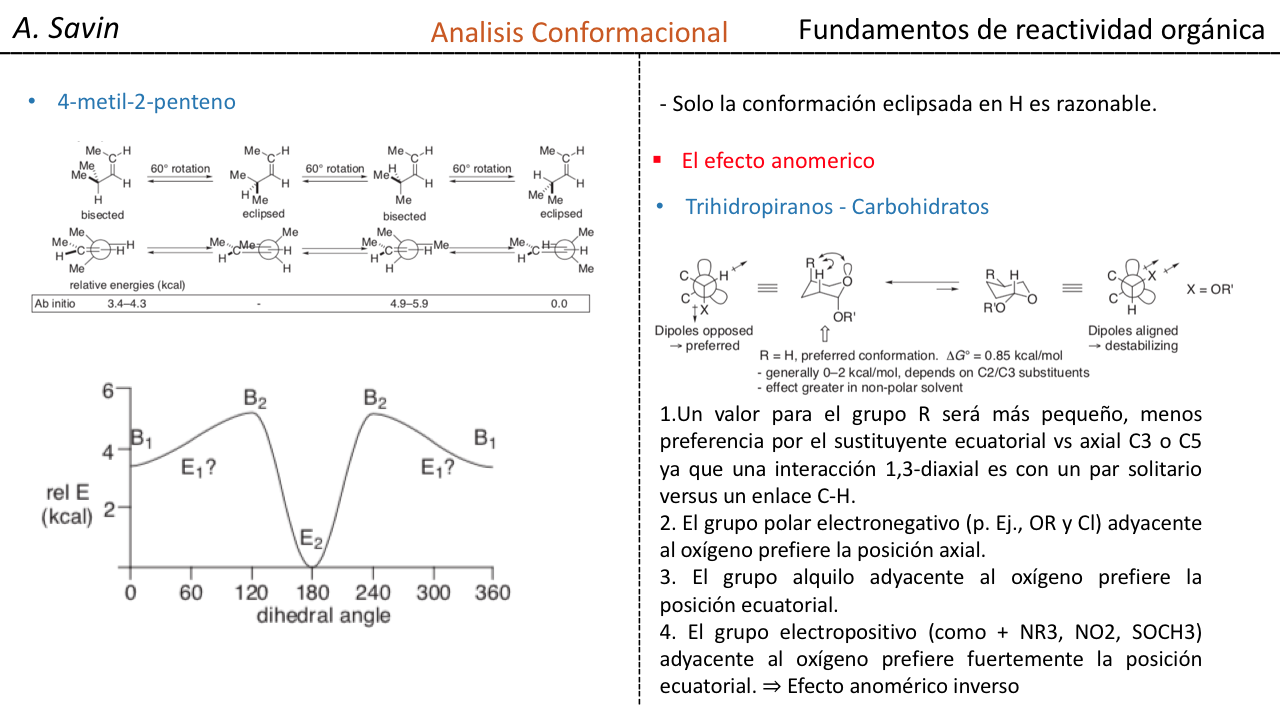
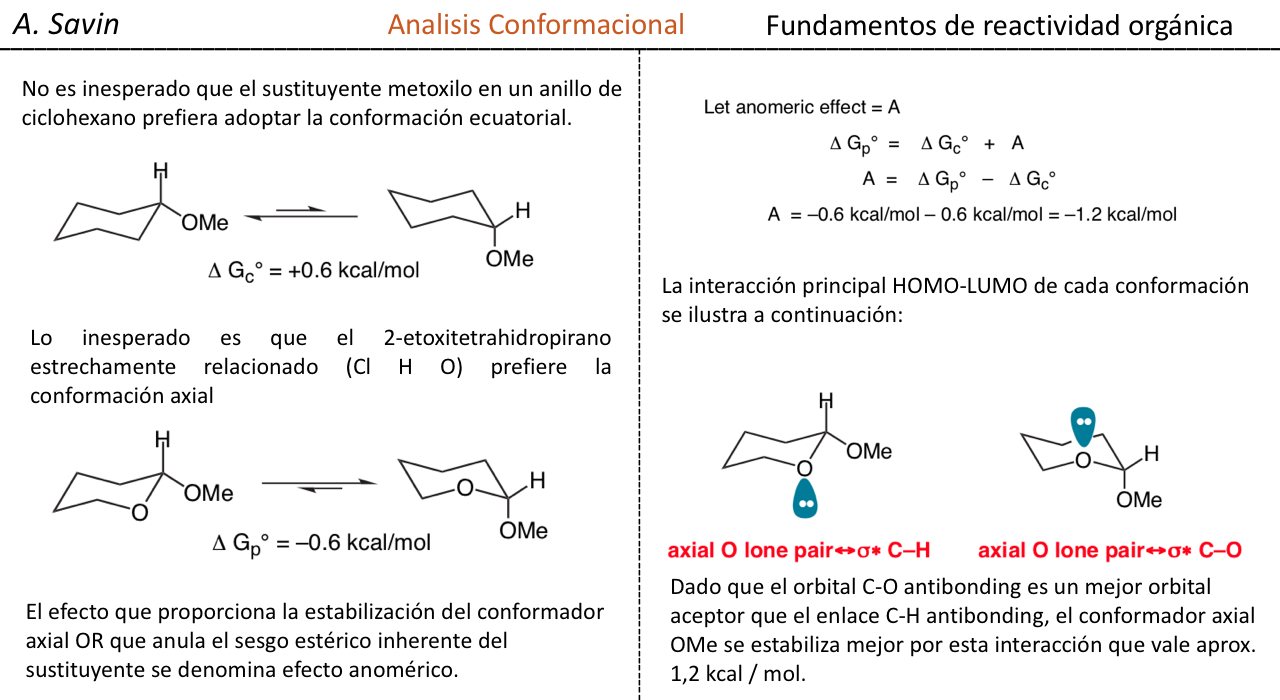
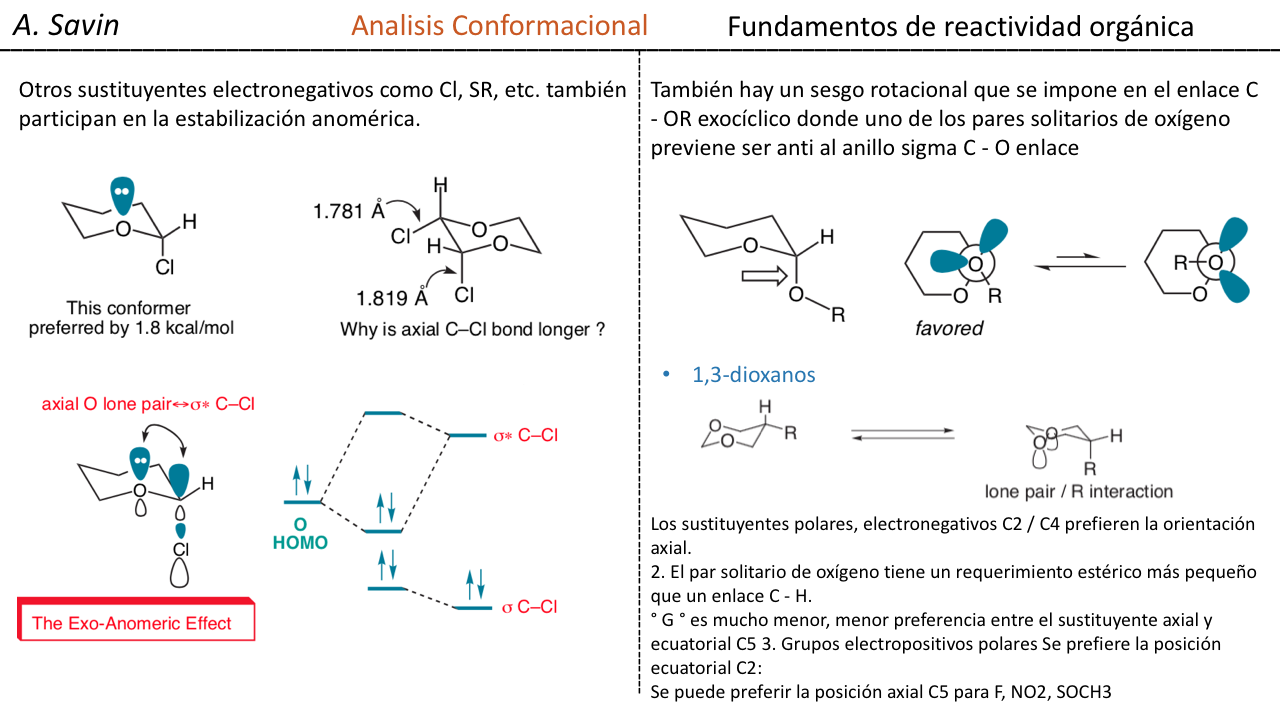
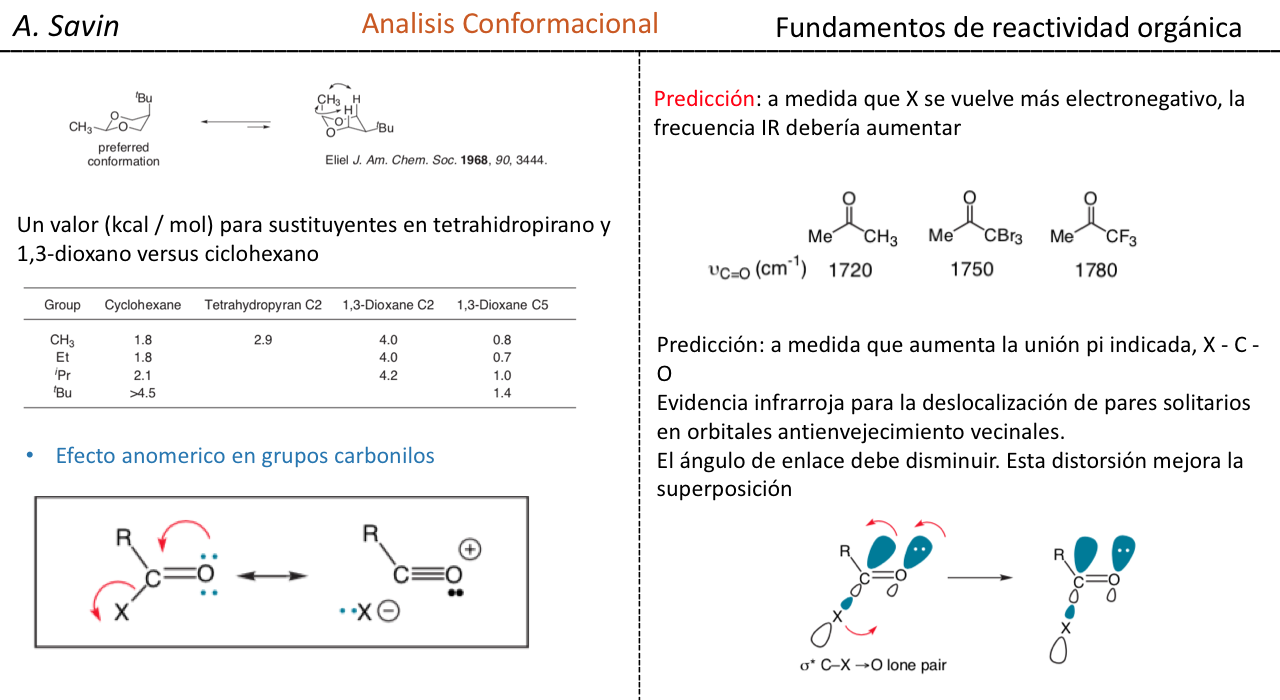
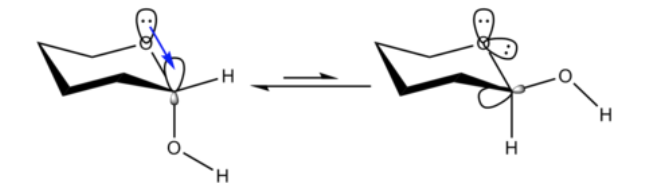
No es inesperado que el sustituyente metoxilo en un anillo de ciclohexano prefiera adoptar la conformación ecuatorial.
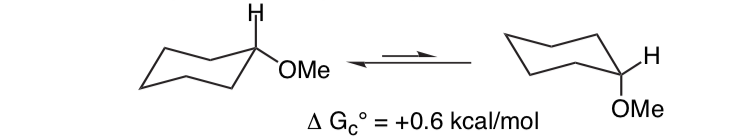
Lo que es inesperado es que el 2-metoxitetrahidropirano estrechamente relacionado Cl H O Prefiere la conformación axial:
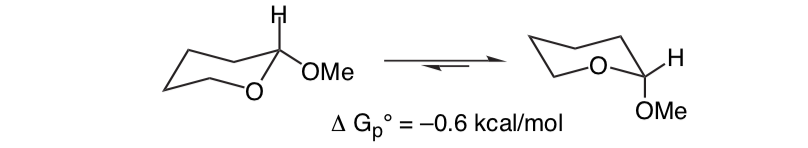
Este efecto que proporciona la estabilización del conformador axial O que anula el sesgo estérico inherente del sustituyente se denomina efecto anomérico.
De la siguiente manera quedaria la relacion de la energia libre respecto al efecto anomerico:
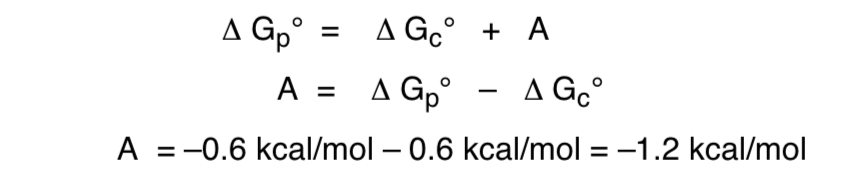
Fuentes recomendadas para expandir la busqueda :
1) Juaristi E. “Conceptos básicos de la teoría orbital”. CINVESTAV, México, 1988 Juaristi E. “Fisicoquímica Orgánica”. CINVESTAV, México, 1994.
2) Neil S. I. “Physical Organic Chemistry” Longman, Milan, 1995.
3) March J., “Advanced Organic Chemistry” John Wiley & Sons, New York, 1992 4) Jones R. A. Y. “Physical and Mechanistic Organic Chemistry”, 2nd. Ed Cambrige University Press, Cambrige, 1984 .
5) Woodward R. B. y Hoffmann R. “The conservation of orbital symmetry”, Academic Press, New York, 1979.
6) Carpenter B. K. “Determination of Organic Reaction Mechanisms”, John Wiley & Sons, New York, 1984 .
- Detalles
- Alejandro Savin.
- Visto: 42833
Estereoquímica
La estereoquímica es el estudio de los compuestos orgánicos en el espacio. Para comprender las propiedades de los compuestos orgánicos es necesario considerar las tres dimensiones espaciales. Las bases de la estereoquímica fueron puestas por Jacobus van’t Hoff y Le Bel, en el año 1874, asi como por Ernest L. Eliel en el siglo XX . De forma independiente propusieron que los cuatro sustituyentes de un carbono se dirigen hacia los vértices de un tetraedro, con el carbono en el centro del mismo.
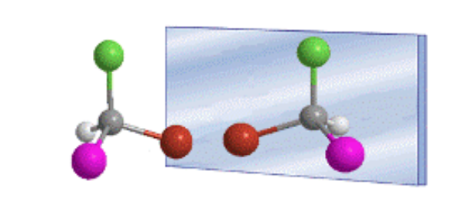
La disposición tetraédrica de los sustituyentes de un carbono sp3da lugar a la existencia de dos posibles compuestos, que son imágenes especulares no superponibles, llamados enantiómeros.
En general a las moléculas que se diferencian por la disposición espacial de sus átomos, se les denomina estereoisómeros.
Isomeria
Se denominan isómeros a aquellos compuestos que tienen idénticas fórmulas moleculares pero que se diferencian en la naturaleza o ordenación de los enlaces entre sus átomos o en la disposición de sus átomos en el espacio.
Para interpretar las diferencias en la propiedades, los químicos imaginaron, el siglo pasado, que los átomos de una molécula tenían disposiciones espaciales concretas que justificaban sus diferentes comportamientos.
La clasificación por función química, establecida según el comportamiento de los compuestos, se ha relacionado con la presencia en la molécula de un grupo de átomos llamado grupo funcional.
Además de la importancia del grupo funcional, existe una diferencia de comportamiento inducida por ligeras diferencias en la disposición de los diferentes átomos que forman el resto de la molécula. Estas diferencias pueden responder a distintas clases de isomerías :
Isomería de función
Pertenecen a este tipo de isomería los isómeros constitucionales, que se diferencian unos de otros en que sus grupos funcionales son distintos.
El grupo funcional en ambos isómeros es distinto
C2H6O
Etanol (CH3-CH2-OH) y dimetiléter (CH3-O-CH3)
El alcohol reacciona con el sodio mientras que con el éter no se observa ninguna reacción.
Del punto de vista físico, el alcohol es un líquido con una temperatura de ebullición de 78,5°C, mientras que el éter es un gas que se licua a -23°C.
Isomería de posición y/o de esqueleto.
Los grupos funcionales son idénticos pero están colocados en posiciones distintas del esqueleto molecular (isómeros de posición).
Ej: 2-hexanol y 3-hexanol :
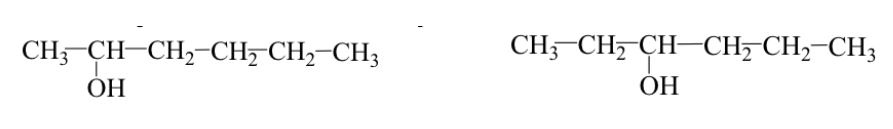
A veces el grupo alquilo tiene una disposición distinta (isómeros de esqueleto o de ramificación).
Ej: 3-metil-2-pentanol y 2-hexanol
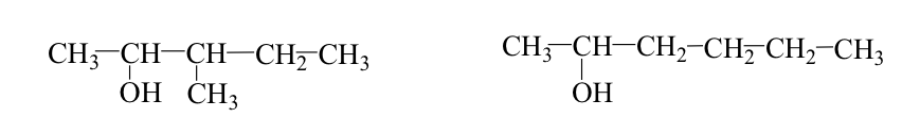
Se pueden producir los dos casos simultáneamente:
Ej : 3-metil-2-pentanol y 3-hexanol
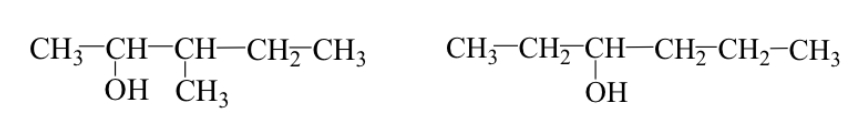
Tautomería
Son isómeros constitucionales de fácil interconversión por encontrarse entre si en rápido equilibrio. El fenómeno recibe el nombre de tautomería y suele consistir en que un átomo, generalmente de hidrógeno, situado en una triada de átomos, y un doble enlace cambian de posición simultáneamente.
El ejemplo más clásico es el equilibrio ceto-enólico (-enopara el doble enlace y -olpara el alcohol).
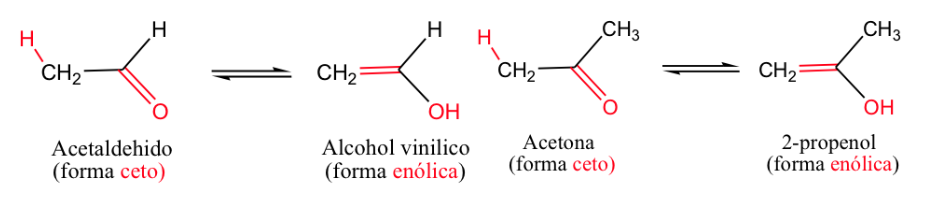
Generalmente las formas cetónicas son las más estables, pero cuando la forma enólica se estabiliza (por enlace hidrógeno o por resonancia) el equilibrio se desplaza.
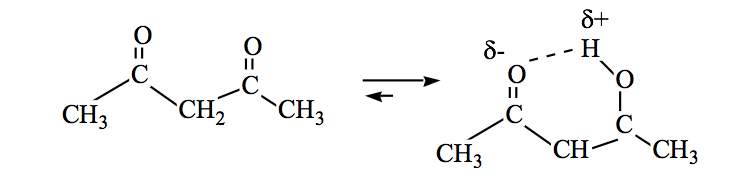
Las amidas también pueden estar en equilibrio ceto-enólico:
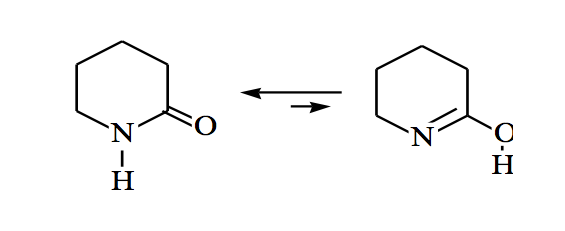
Tautomería imina-enamina:
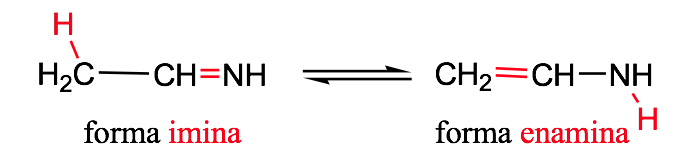
nitro-aci
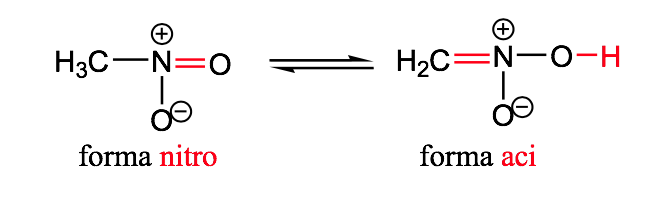
Isomería geométrica
Estas dos formas no son isómeros geométricos ya que la libre rotación entorno al enlace simple convierte una forma en otra (confórmeros)
El doble enlace no permite la libre rotación lo que puede generar dos estructuras distintas dependiendo de la posición de los grupos A y B en el espacio: son isómeros geométricos.
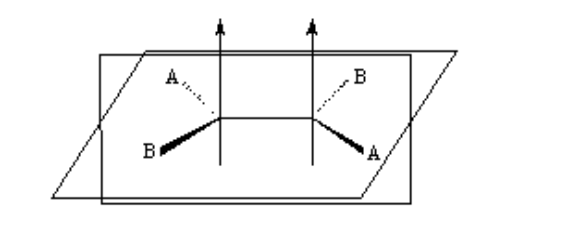
Isomería en los alquenos
Para que exista isomería geométrica se deben cumplir dos condiciones:
1.- Rotación impedida (por ejemplo por un doble enlace)
2.- Dos grupos diferentes (A y B) unidos a ambos lados del enlace
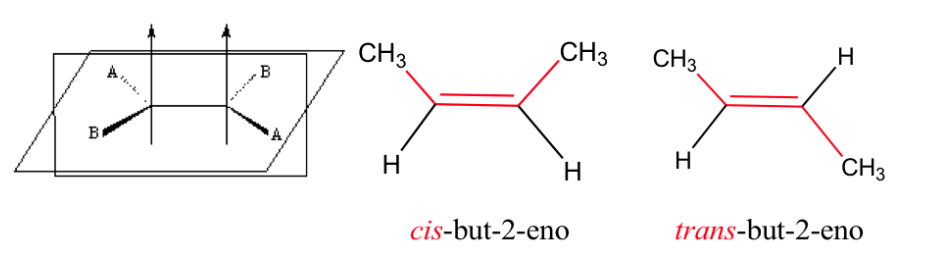
Efecto de la isomería geométrica sobre las propiedades físicas

Nomenclatura de isómeros geómétricos
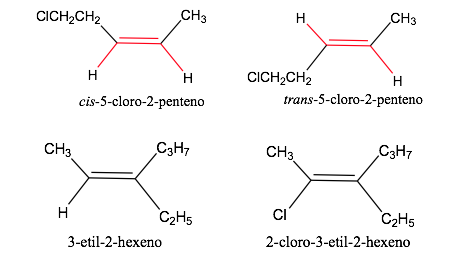 Reglas de secuencia o prioridad
Reglas de secuencia o prioridad
Las reglas que se deben tener en cuenta para establecer el orden de prioridad o preferencia de los átomos o grupos de átomos fueron establecidas en 1956 por Cahn, Ingoldy Prelogy modificadas varias veces para evitar ambigüedades.
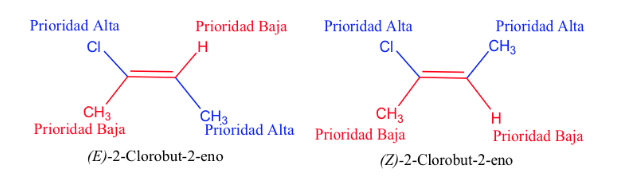
Cuando los grupos de prioridad alta se encuentren en lados opuestos del plano perpendicular a la molécula, el isómero se llama E
Cuando están en el mismo lado de este plano el isómero se denomina Z
Ejemplo : but-2-eno :
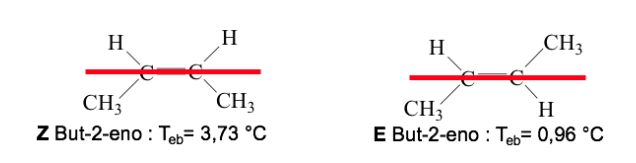
La prioridad de los sustituyentes sobre los carbonos del doble enlace se puede deducir de las siguientes reglas:
- Regla 1:
- Si los átomos unidos al átomo de carbono en estudio son distintos, tienen prioridad los del número atómico más alto sobre los del número atómico más bajo y si se trata de dos isótopos, se consideran por orden decreciente de masa atómica.
Ej : Br (35) > Cl (17) > O (8) > N (7) > C (6) > H (1)
D > H y 13C >12C
Regla 2:
Cuando los átomos unidos al átomo de carbono son idénticos (y no sirve la primera regla), se sigue la secuencia, es decir, se recurre a la comparación de los átomos unidos a ellos y, si es preciso porque fueran también iguales, a los siguientes, etc, teniendo en cuenta que si los átomos son iguales pero en número diferente, tiene prioridad el sustituyente con más átomos de rango superior.
Ej : -CH2-OH > -CH3 porque O > H
-CH2-Br > -CH2-OH porque Br > OH
-CH2-CH3> -CH3 porque C > H
Regla 3:
Los enlaces dobles y triples se tratan como si fueran sencillos, duplicando o triplicando los átomos de la cadena, respectivamente.
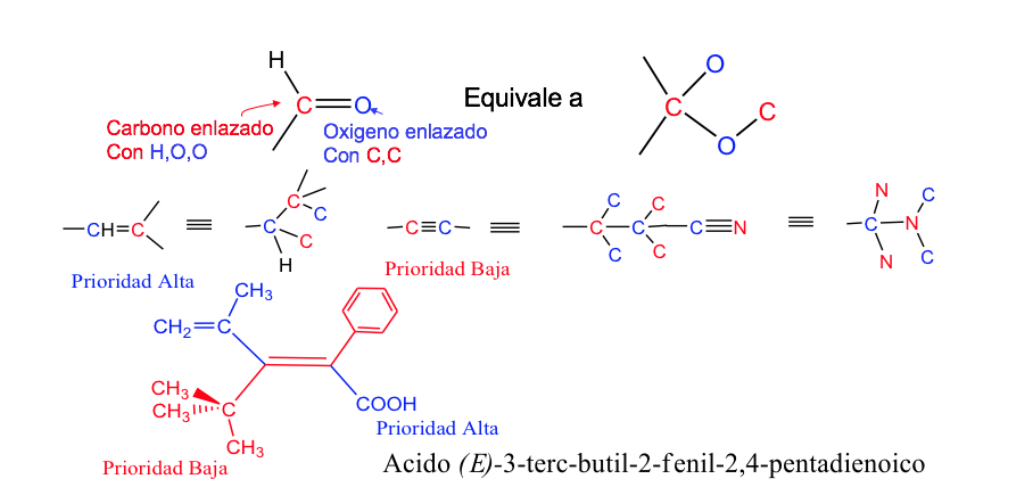
. Isomería de los ciclos y los sistemas complejos.
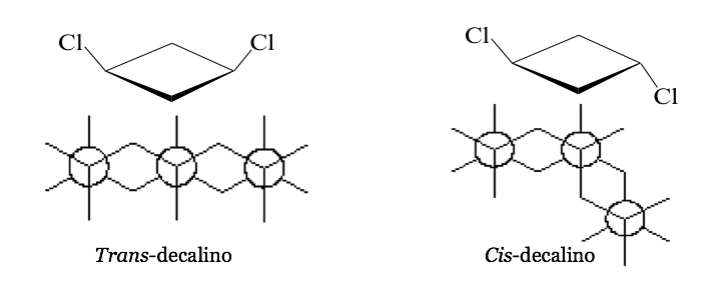
En algunas moléculas cíclicas simétricas (al menos disustituidas), los átomos del ciclo definen un plano. Un sustituyente se sitúa hacia una cara de este plano mientras que el otro puede estar situado hacia el mismo lado o hacia el lado opuesto.
Cis-1,3-Diclorociclobutano trans-1,3-Diclorociclobutano
Representación proyectiva (CRAM)
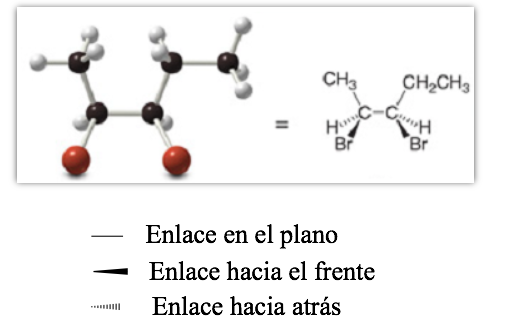
Representación en perspectiva
Representación de Newman
Una proyección de Newman es una forma de representación bidimensional útil para visualizar conformaciones en un enlace simple carbono-carbono de una molécula orgánica.
Representación de Fischer.
La proyección de Fisher es una forma estándar de dibujar en dos dimensiones átomos de carbono tetraédricos y sus sustituyentes.
En esa proyección cada carbono tetraédrico se representa como una cruz en la que, las líneas horizontales se dirigen hacia afuera del papel y las verticales hacia adentro.
La representación de Fisher se puede hacer marcando los enlaces que estén delante del plano con trazo grueso y los situados detrás con trazo discontinuo, pero generalmente los diferentes enlaces se presentan con trazos normales, aunque se sobre entiende que los sustituyentes representados a derecha e izquierda de la línea vertical están por encima del plano de representación y los representados arriba y abajo, por debajo de ese plano.
Por convención general la cadena carbonada se presenta en la vertical poniendo el carbono más oxidado en la parte superior.
Representación de Harwoth
- Es una representación en perspectiva de las formas cíclicas de las moléculas de azúcar de 5 o 6 átomos (furanosas, piranosas).
- Ej. :
Quiralidad
Se denomina quiral toda figura geométrica, o todo grupo de puntos, cuya imagen en un espejo plano, idealmente realizada, no puede hacerse coincidir consigo misma. Algunas moléculas son como las manos. La izquierda es la imagen especular de la derecha pero no son superponibles y por lo tanto no son idénticas. Se llaman quirales.
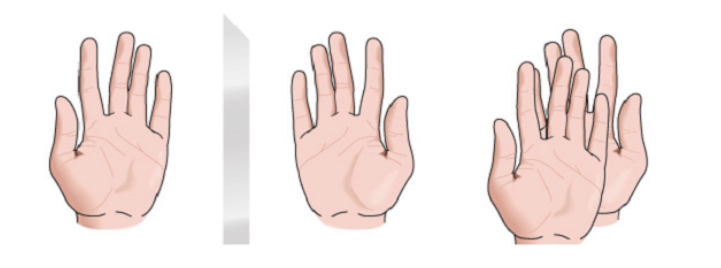
Hay otras moléculas parecidas a un par de calcetines. Los calcetines son imágenes especulares un del otro y también son superponibles.
Una molécula o un objeto superponible a su imagen especular se denomina aquiral.
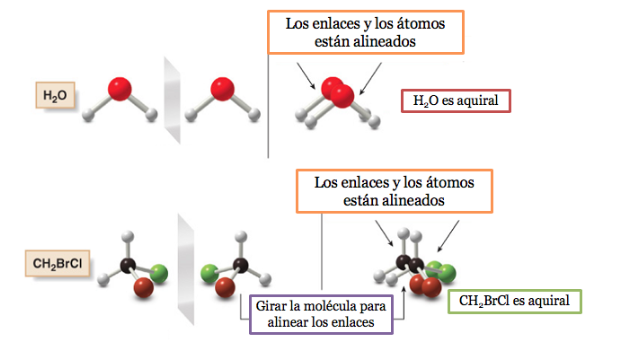
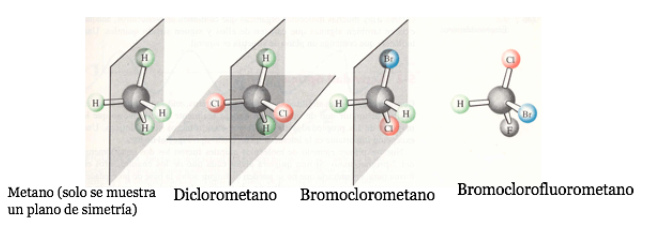
Si una molécula posee un plano de simetría es un sistema aquiral.
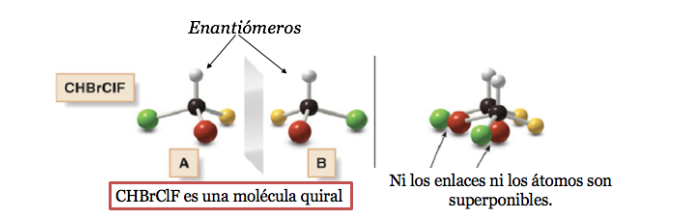
Una molécula quiral existe en dos formas estereoisómericas llamadas enantiómeros. Estos son objetos no superponibles con sus imágenes especulares.
Un átomo de carbono unido a 4 sustituyentes distintos se denomina carbono asimétrico. Sin embargo su existencia no es garantía de quiralidad (como se verá más adelante). Se llama también carbono estereogénico o estereocentro.
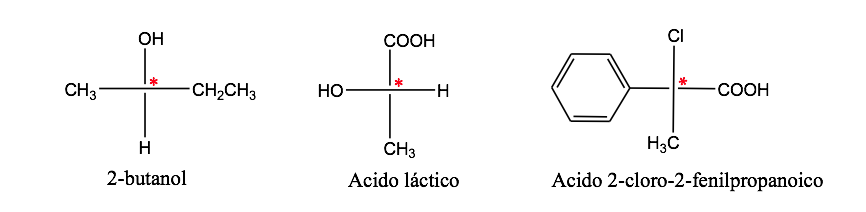
Isomería óptica
Las propiedades físicas de dos enantiómeros son idénticas: tienen los mismos puntos de ebullición y de fusión, la misma solubilidad, la misma densidad, el mismo índice de refracción, misma conductividad...etc.
La actividad óptica de las parejas de enantiómeros es la propiedad característica para diferenciarlos.
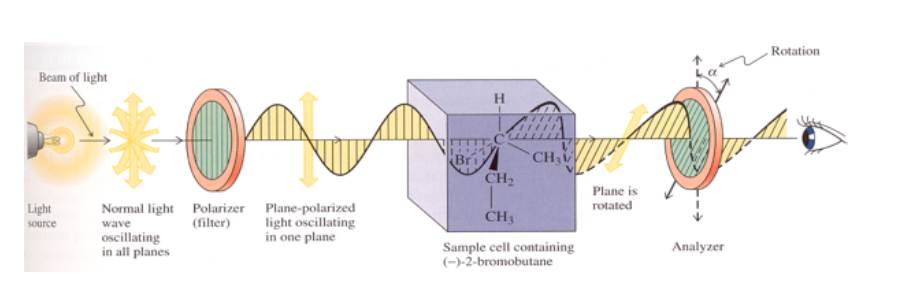
Si la sustancia no es ópticamente activa no se observa ningún cambió en el plano de vibración de la luz polarizada emitida.
Si la sustancia tiene actividad óptica se observa una rotación de agrados del plano de vibración de la luz polarizada emitida.
Si la rotación del plano de la luz es hacia la derecha (en el mismo sentido de las agujas del reloj) la sustancia es dextrógira y al valorα se le asigna signo positivo.
Si el giro es hacia la izquierda (en el sentido contrario de las agujas del reloj) la sustancia es levógira y a αse le asigna signo negativo.
La rotación específica de una molécula ópticamente activa es una constante física característica de dicha molécula

Rotación específica de algunos compuestos quirales
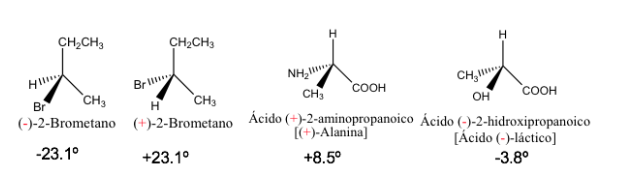
Los enantiómeros puros presentan el mismo valor de rotación específica pero de signo contrario.
Por lo tanto, la rotación óptica resultante de una mezcla 1:1 de enantiómeros es cero, es decir, es ópticamente inactiva. Este tipo de mezclas se denomina racematoo mezcla racémica.
La denominación de la configuración absoluta de un centro estereogénico se basa en la mismas reglas de prelación desarrolladas por Cahn, Ingold y Prelog.
Estas reglas permiten nombrar y describir la disposición en el espacio de los sustituyentes sobre un centro estereogénico, independientemente del signo de la rotación óptica de la molécula.
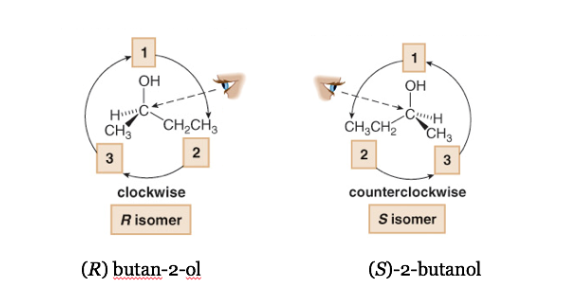
El sustituyente de menor prioridad se encuentra lo más alejado posible del observador.
Si el paso de 1à2à3 se hace en el sentido horario, el centro quiral es R(rectus, latín, derecha).
Si el paso 1à2 à3 se hace en el sentido contrario a las agujas del reloj, la configuración del centro quiral se denomina S(sinister, latín, izquierda).
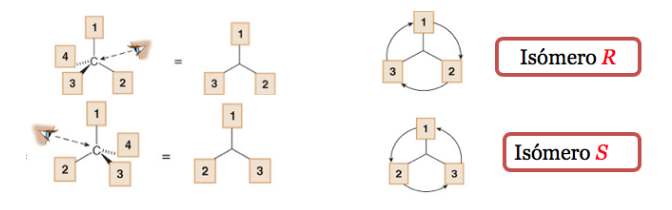
En la nomenclatura sistemática, R o Sse añaden entre paréntesis como prefijo del nombre del compuesto quiral
Es importante recordar que los símbolos R y S no muestran ningún tipo de correlación con el signo de α.
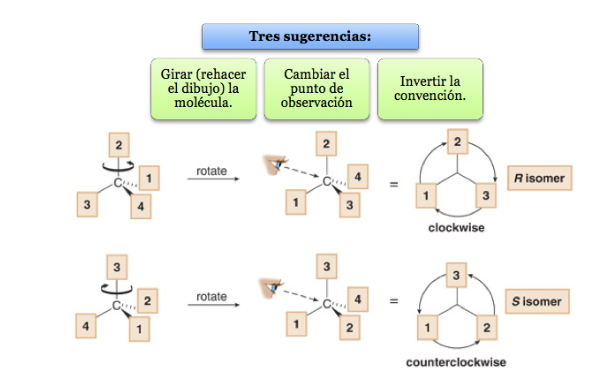
¿Que hacer cuando una molécula no está orientada de manera que el grupo de menor prioridad esté alejado?
Un compuesto con ncentros estereogénicos tiene un máximo de 2n estereoisómeros.
Ejemplo:
Un compuesto con dos centros estereogénicos tiene un máximo de 4 estereoisómeros.
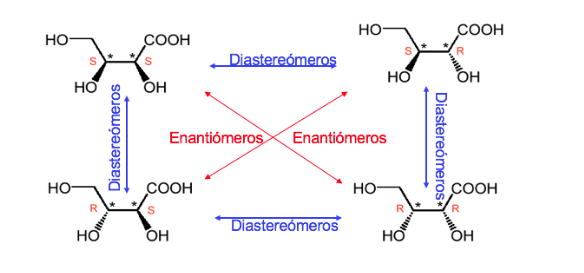
Se llama forma o compuesto meso a todo esteroisómero cuya molécula no es quiral a pesar de poseer centros estereogénicos
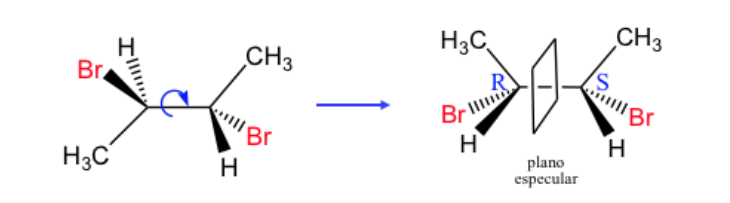
Un compuesto con dos carbonos asimétricos se domina like cuando los dos carbonos tienen la misma configuración absoluta y unlike en el caso contrario.
Compuestos eritro y treo
Cuando dos carbonos tienen al menos dos sustituyentes idénticos se puede usar la denominación treoy eritro.
Un par de enantiómeros eritro es aquel en el que los grupos idénticos se pueden poner en posición eclipsada.
No hay una relación directa entre la nomenclatura Ry Sy la nomenclatura Eritro/Treo .
Cuando dos carbonos tienen tres sustituyentes idénticos la forma eritro es mesoya que presenta un plano de simetría.
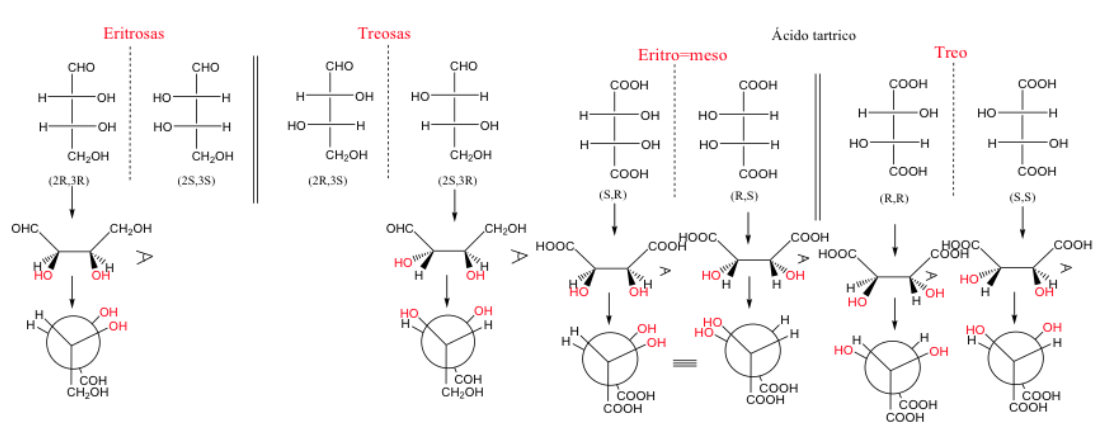
Una molécula recibe el nombre de eritrocuando, en su representación de Fischer, los grupos iguales o parecidos están al mismo lado.
Un molécula es treo si estos grupos están en los lados opuestos.
(usada generalmente para los azúcares (osas))
Es una nomenclatura anterior a la nomenclatura Ry S.
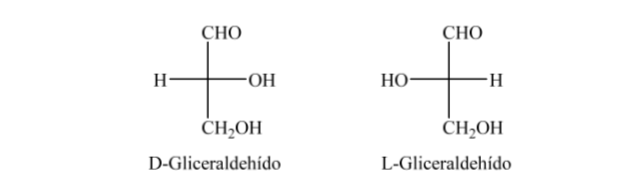
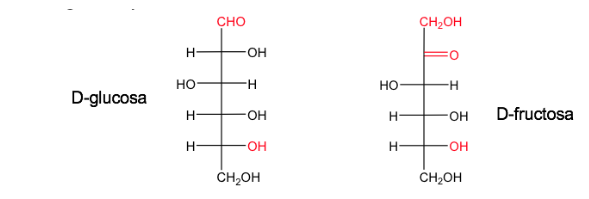
Un azúcar es nombrado D cuando, en la proyección de Fischer (con el carbono más oxidado situado arriba)el hidroxilo asociado al carbono asimétrico de mayor numeración queda a la derecha.
Su enantiómero se llamara Ly tendrá el OH equivalente a la izquierda.
La glucosa y la fructosa en sus formas naturales existen como D.
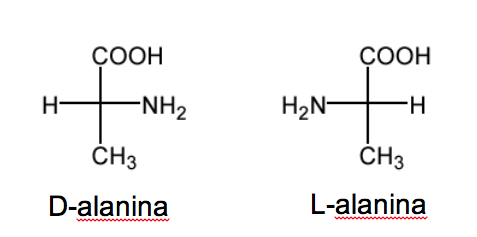
La nomenclatura D/L se emplea también en la serie de los aminoácidos
RCH(NH2)COOH.
En los azúcares esta nomenclatura depende de la posición del hidroxilo. En este caso es la posición del grupo amino la que define la nomenclatura. Cuando en la proyección de Fisher (con el carbono más oxidado arriba) el grupo NH2 está en la derecha el etereoisómero es D y su enantiómero es L.
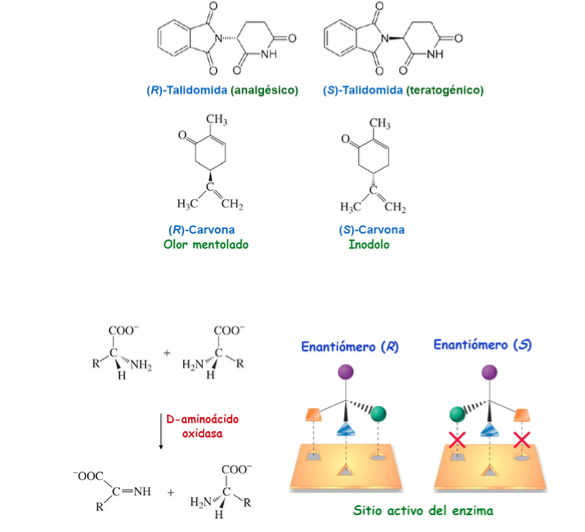
Importancia de la quiralidad
Fuentes para expandir la busqueda de conocimiento:
1) Juaristi E. “Introduccion a la estereoquimica y al analisis conformacional”. CINVESTAV, México, 1988 Juaristi E. . CINVESTAV, México, 1994.
2) Neil S. I. “Physical Organic Chemistry” Longman, Milan, 1995.
3) March J., “Advanced Organic Chemistry” John Wiley & Sons, New York, 1992 4) Jones R. A. Y. “Physical and Mechanistic Organic Chemistry”, 2nd. Ed Cambrige University Press, Cambrige, 1984 .
5) Woodward R. B. y Hoffmann R. “The conservation of orbital symmetry”, Academic Press, New York, 1979.
6) Carpenter B. K. “Determination of Organic Reaction Mechanisms”, John Wiley & Sons, New York, 1984 .
- Detalles
- Alejandro Savin.
- Visto: 15033
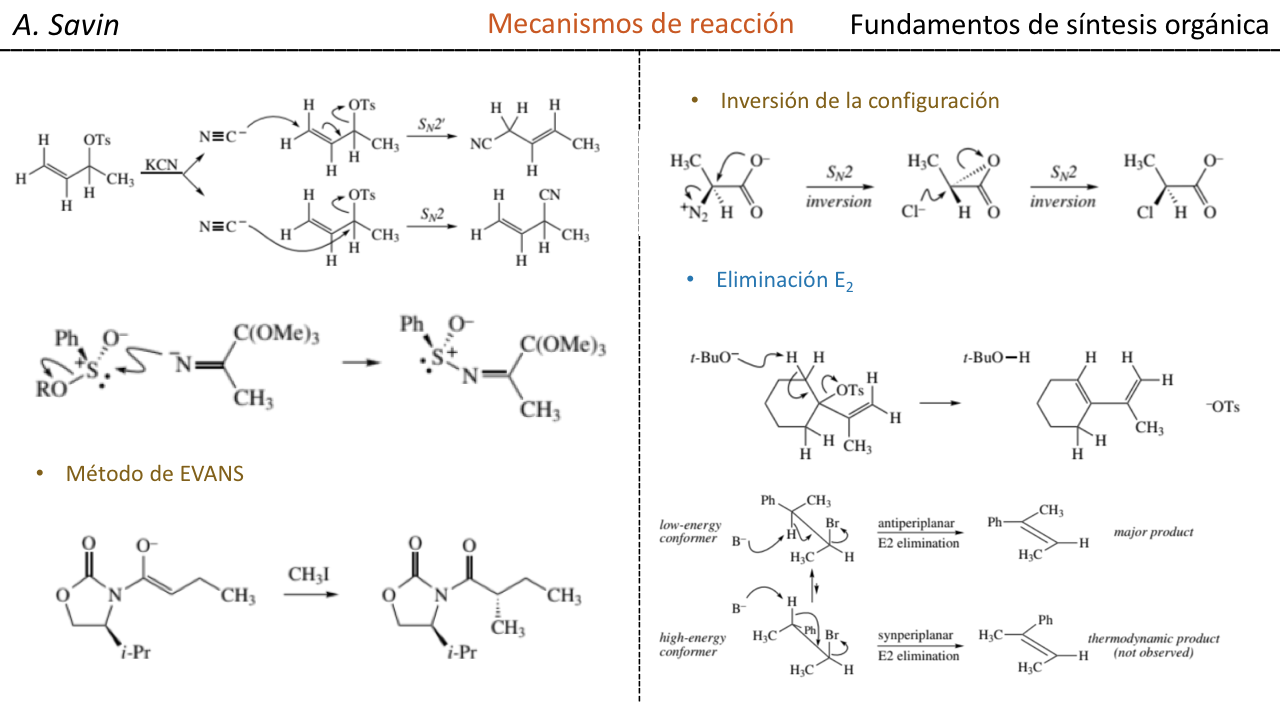
Enestereoquímica, la inducción asimétrica(tambiénenantio-inducción) en unareacción químicadescribe la formación preferencial de unenantiómeroodiastereómerosobre el otro, como resultado de la influencia de una característicaquiralpresente en elsustrato,reactivo,catalizadoro ambiente. La inducción asimétrica es un elemento clave en lasíntesis asimétrica.
La inducción asimétrica fue introducida porEmil Fischer, basado en su trabajo sobre loscarbohidratos. Existen varios tipos de inducción.
Lainducción asimétrica internahace uso de un centro quiral unido al centro reactivo por medio de unenlace covalente, y permanece así durante la reacción. En lainducción asimétrica por relayla información quiral es introducida en un paso separado, y eliminado nuevamente en una reacción química separada. Los sintones especiales son denominadosauxiliares quirales. En lainducción asimétrica externa, la información quiral es introducida en elestado de transicióna través de uncatalizadoroligando quiral. Este método desíntesis asimétricaes económicamente el más deseable.
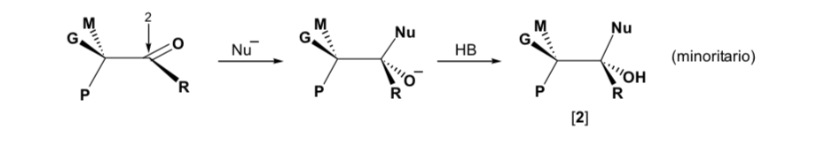
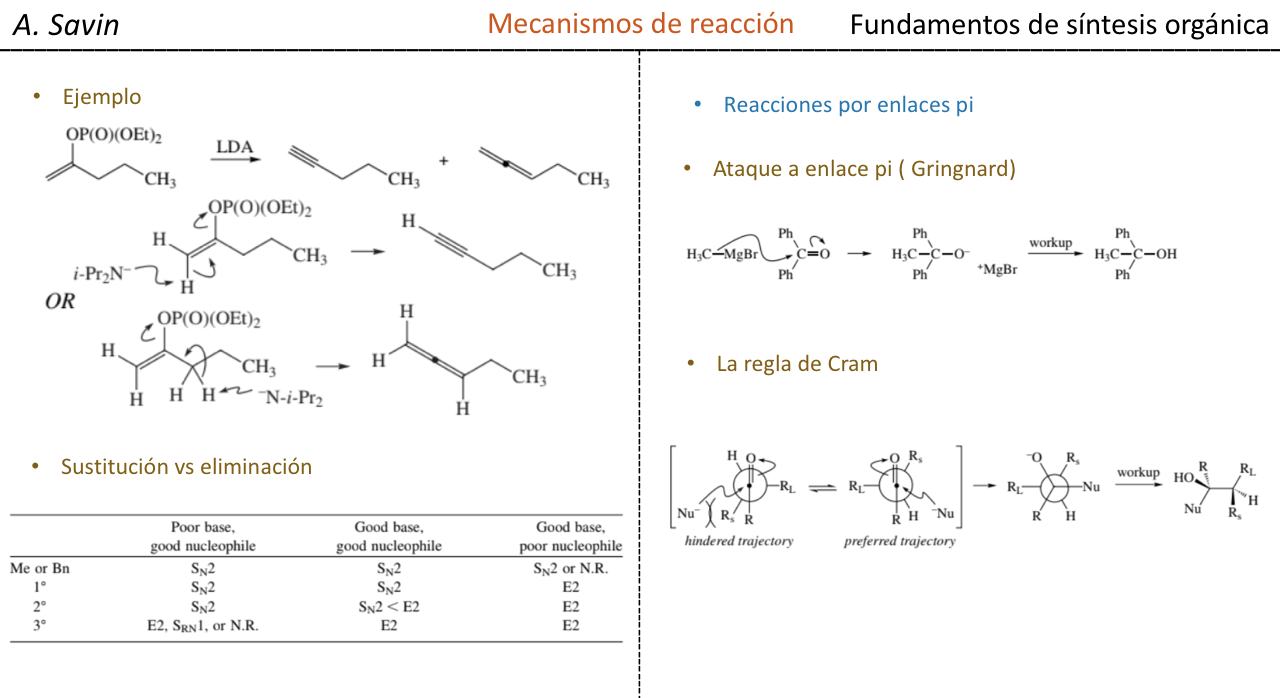
Estereoselectividad en la reaccion de adicion nucleofilica a carbonilos (Reglas de Cram)
En las reacciones de adición nucleófila, el átomo de carbono del grupo carbonilo puede transformarse en un átomo de carbono asimétrico, dependiendo del tipo de nucleófilo empleado y de los radicales alquilo que están unidos inicialmente al C=O.
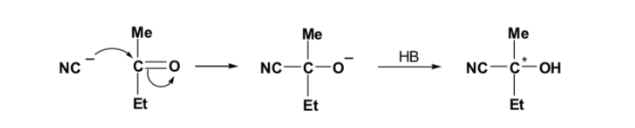
Si el compuesto carbonílico y el nucleófilo no son quirales se obtiene una mezcla equimolecular de dos enantiómeros (racémico):
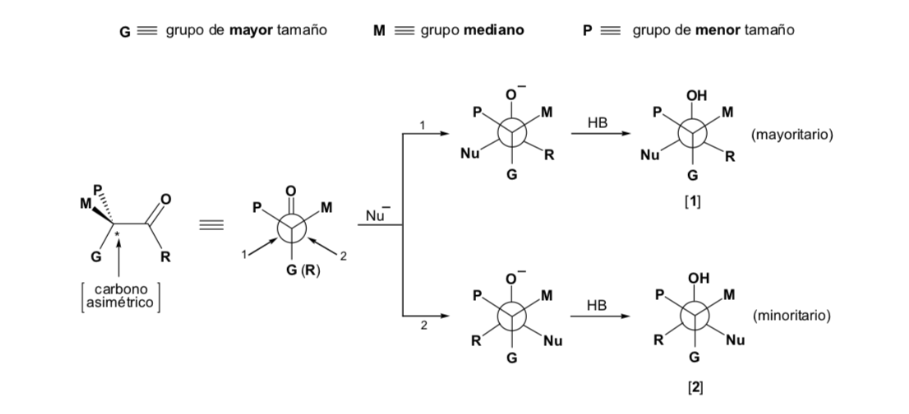
Sin embargo, cuando el compuesto carbonílico es quiral, la probabilidad de que el nucleófilo reaccione por cada una de las dos caras del grupo carbonilo no es la misma. El impedimento estéreo hace que la reacción por una de las caras esté favorecida, y el resultado es una mezcla de diastereoisómeros en proporciones diferentes (inducción asimétrica):
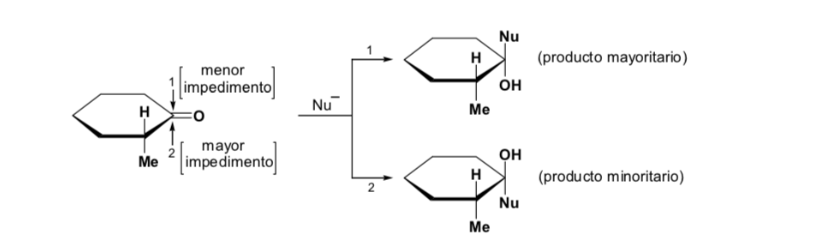
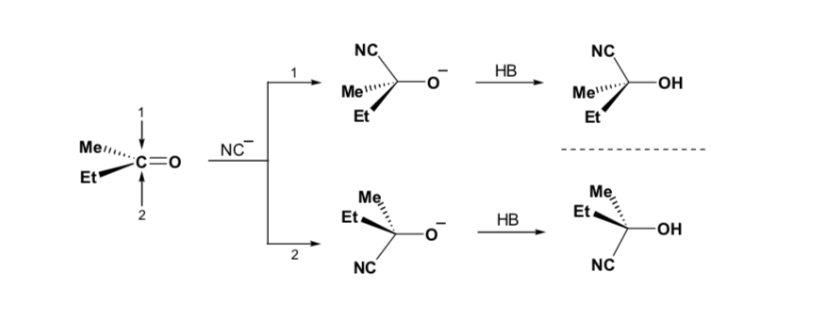
El diastereoisómero mayoritario se forma cuando el nucleófilo reacciona con el grupo carbonilo por el lado menos impedido, y la conformación del sustrato es aquella en la que el grupo carbonilo está flanqueado por los grupos menos voluminosos unidos al Cα asimétrico.
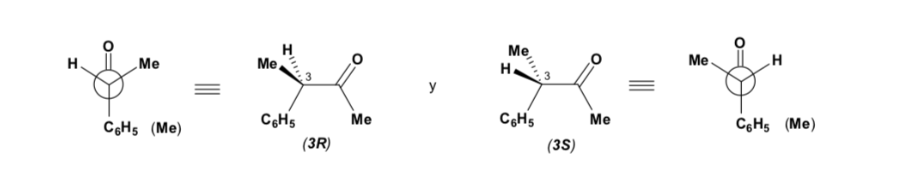
[grupo carbonilo flanqueado por los grupos menos voluminosos: H y Me]
La regla de Cram se refiere a la reacción de uno de los estereoisómeros que forman la pareja de enantiómeros, no a la reacción del racémico con el nucleófilo.
El nucleófilo se aproxima por el lado en que están situados P y G:
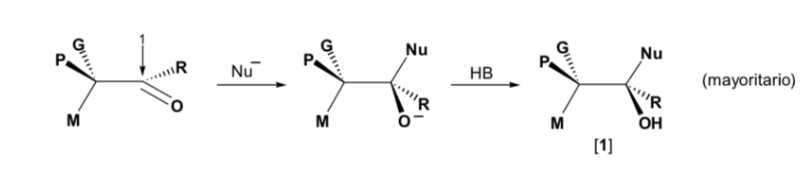
El nucleófilo se aproxima por el lado en que están situados M y G:
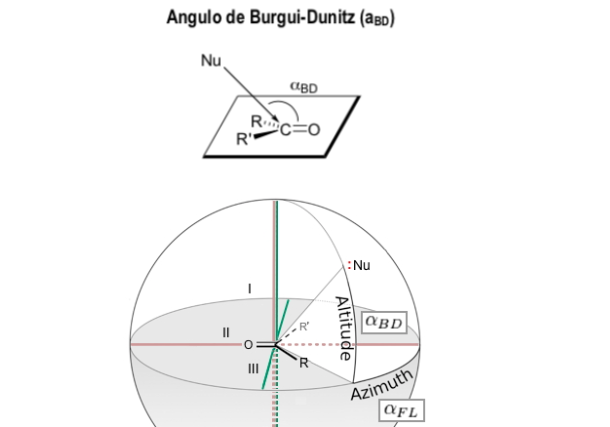
Angulo de Bürgi-Dunitz
El ángulo Bürgi-Dunitz (ángulo BD) es uno de los dos ángulos que definen completamente la geometría de "ataque" (aproximación por colisión) de un nucleófilo en un centro trigonal insaturado en una molécula, originalmente el centro carbonilo en una cetona orgánica, pero ahora se extiende a los carbonilos de aldehído, éster y amida, y también a los alquenos (olefinas).
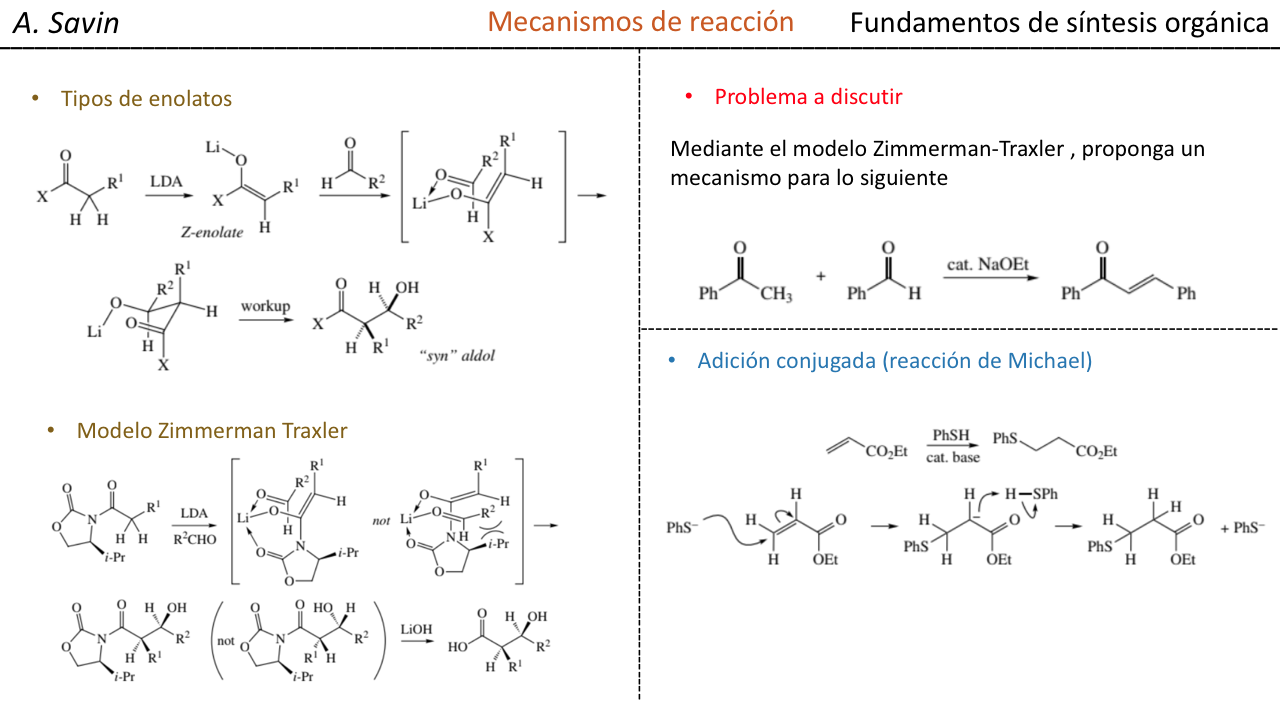
Modelo de zimmerman - traxler
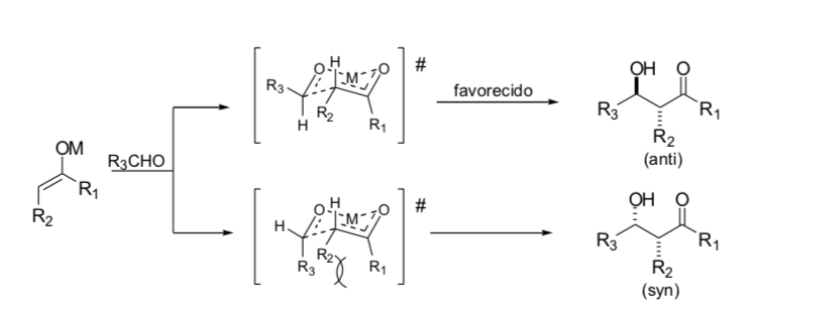
Zimmerman y Traxler propusieron que la reacción aldólica con enolatos metálicos se produce a través de un proceso pericíclico similar a una silla. En la práctica, la estereoquímica puede ser altamente dependiente del metal. Solo unos pocos metales, como el boro, siguen de manera confiable las vías indicadas.
Tension alilica
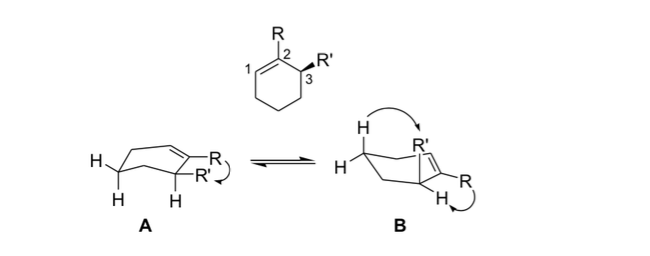
En estereoquimica la tensión alílica o tensión 1,3-alílica es una energía de tensión que resulta de una conformación molecular desfavorable para el grupo alilo, producto de la interacción entre un sustituyente en un extremo de una olefina con un sustituyente alílico del otro extremo.
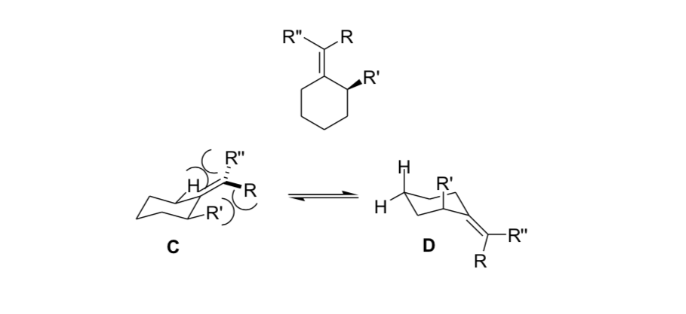
Los químicos orgánicos usan esta rigidez resultante de la tensión alílica para obtener reacciones asimétricas.
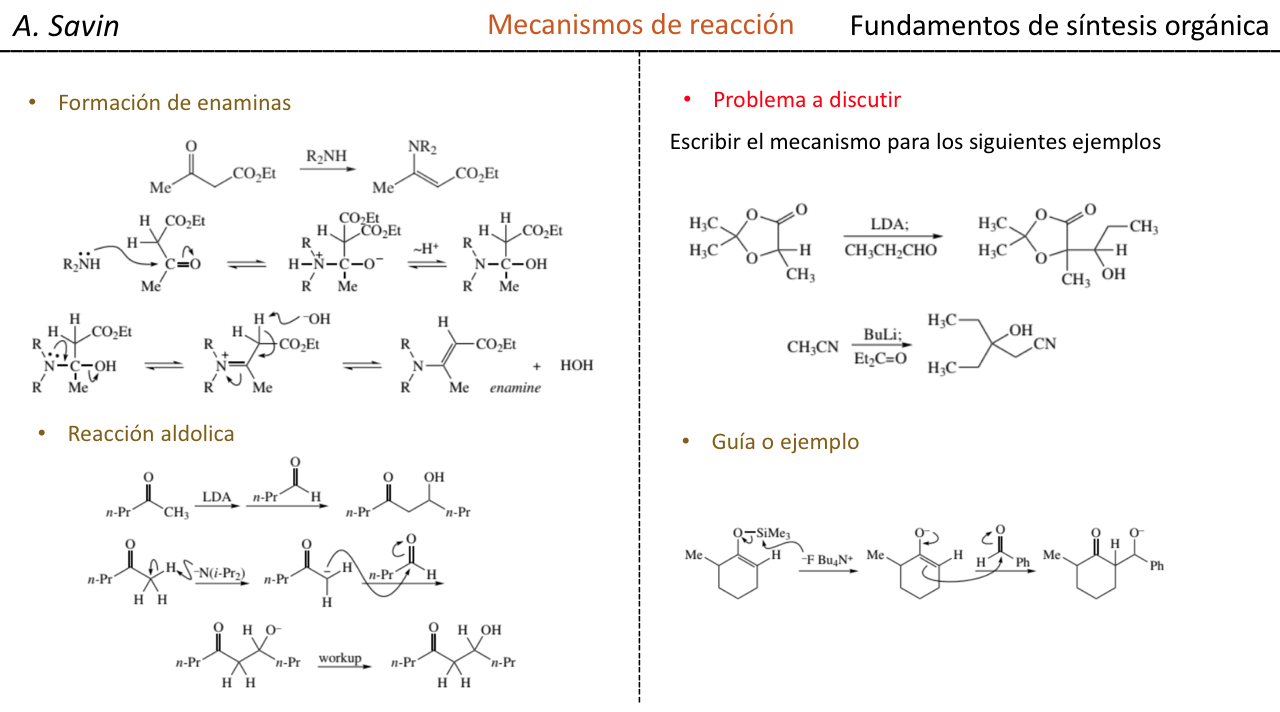
Substitución en la posición alfa de enolatos: introducción de un centro estereogénico nuevo :
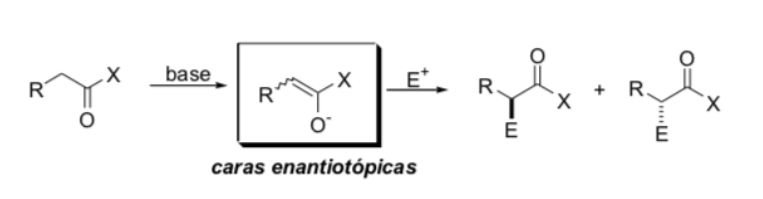
Factores que controlan la estereoselectividad
a) geometría del enolato
b) fuentes de quiralidad presentes ya sea en el enolato o en el electrofilo (fuente de información asimétrica)
c) efectos estereoelectrónicos
Fuentes para expandir informacion :
1) Guo-Qiang Lin, Yue-Ming Li, Albert S. C. Chan. Principles and Applications of Asymmetric Synthesis. Ed. Wiley-Interscience. Great Britain, 2001.
2) Mark Rizzacasa and Michael Perkins. Stoichiometric Asymmetric Synthesis. Ed. Sheffield Academic Press. U.S.A and Canada. 2000.
3) Jonathan M. J. Williams. Catalysis in Asymmetric Synthesis. Ed. Sheffield Academic Press. U.S.A and Canada. 1999.
4) R. A. Aitken and S.N. Kulényi. Asymmetric Synthesis. Ed. Blackie Academic and Professional. Great Britain, 1992.
5) Grossman R.B. The Art of Writing Reasonable Organic Reaction Mechanisms. Springer, New York. 2003
6) Norman R.O.C.; Coxon J. M. Principles of Organic Synthesis. CRC Press, Boca Ratón. 1993
- Detalles
- Alejandro Savin.
- Visto: 13593
Organocatálisis asimétrica
Introducción
A pesar de la importancia de la quiralidad, la obtención de moléculas quirales en forma enantioméricamente pura ha permanecido extremadamente limitada hasta muy recientemente. Desde finales del siglo XIX, la síntesis de moléculas quirales de forma estereoselectiva ha constituido un reto sintético de gran magnitud al que los químicos orgánicos han respondido con gran ingenio y brillantez.
En grandes líneas se pueden considerar tres estrategias básicaspara la obtención de compuestos enantioméricamente puros, como son
a) la resolución de racematos,
b) la utilización de moléculas naturales ópticamente activas
c) la síntesis asimétrica.
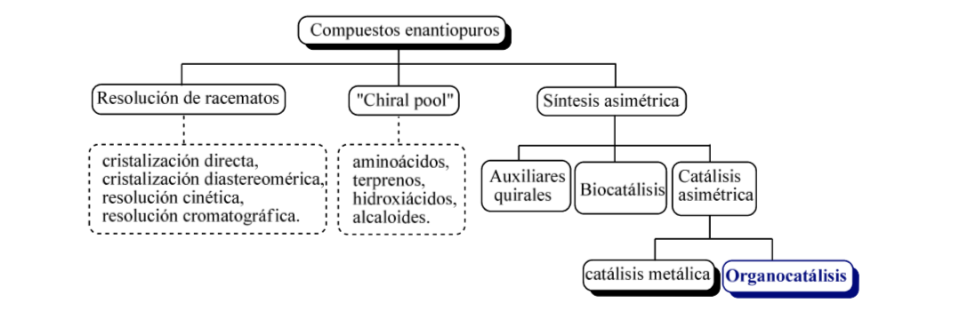
Uso de auxiliares quirales :
Un auxiliar quiral es un compuesto quimico o unidad que se incorpora temporalmente a una síntesis orgánica para que pueda llevarse a cabo asimétricamente, con la formación selectiva de uno de dos enantiómeros
Esta estrategia adquirió un auge considerable en la década de los 80 y en la actualidad se conoce una abundante gama de auxiliares para un gran número de reacciones. Algunos de los auxiliares más representativos se muestran en la figura
¿Qué es la organocatálisis asimétrica?
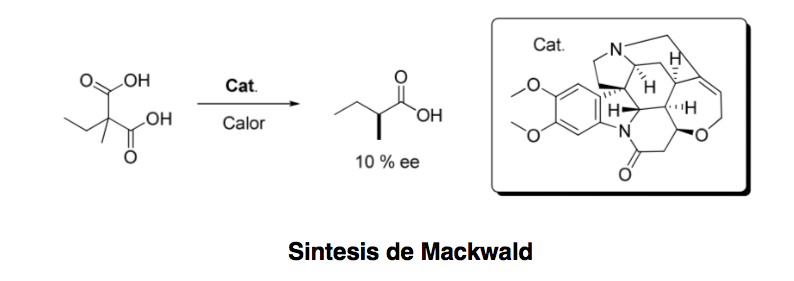
La organocatálisis asimetrica o catalisis asimetrica puede definirse como “la aceleración de reacciones químicas con una cantidad subestequiométrica de un compuesto orgánico, el cual no contiene ningún átomo metálico haciendo uso de moleculas organicas de naturaleza quiral”.Aunque el primer ejemplo de una transformación enantioselectiva organocatalítica data de 1904, cuando Mackwald llevó a cabo la descarboxilación de un derivado de un ácido en presencia de brucina,no ha sido hasta hace unos pocos años cuando este campo ha experimentado un resurgimiento espectacular.
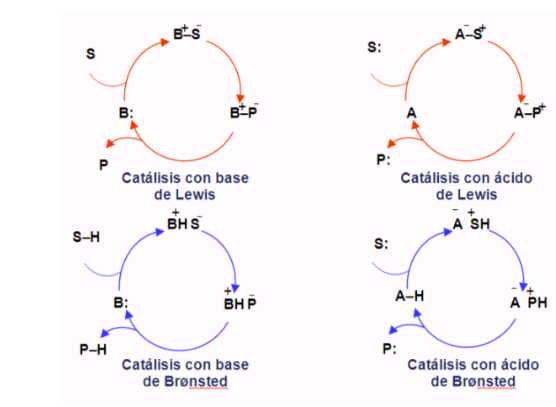
Ciclos organocatáliticos
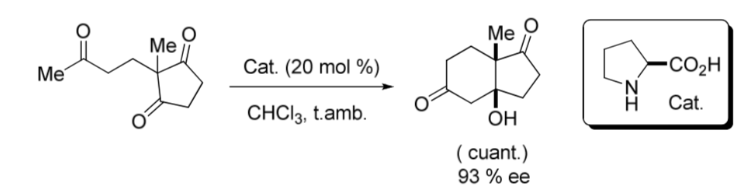
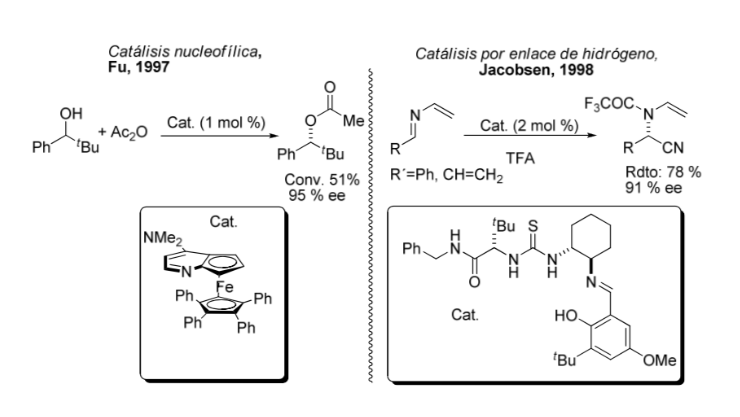
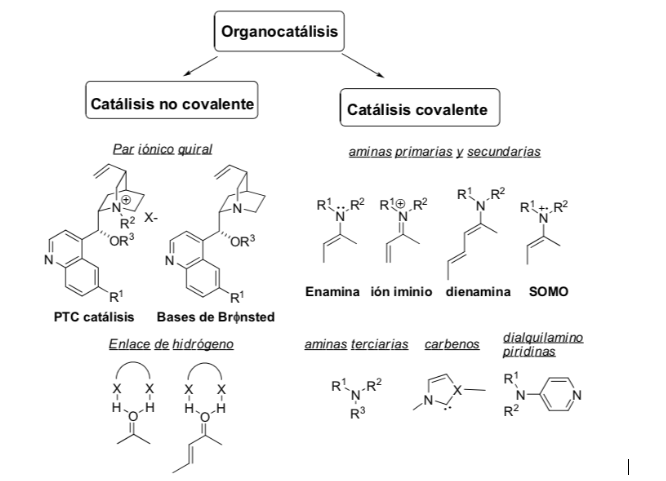
Un hito importante en el área de la organocatálisis asimétrica lo constituyó la reacción aldólica intramolecular catalizada por la L-Prolina publicada a finales de los años 60 y principios de los 70, por los grupos de Hajos y Parrish;y por otro Ender, Wiechert y Sauerindependientemente como se muestra en la siguiente imagen:
Comparacion entre catalisis organometalica y asimetrica:
Método de activación:
Fuentes para expandir la busqueda:
Berkessel, A. y Groeger, H. Asymmetric organocatalysis: from biomimetic concepts to applications in asymmetric synthesis”, Wiley-VCH: Weinheim, 2005.
Bui, T., Syed, S. y Barbas, C.F., Thiourea-Catalyzed Highly Enantio- and Diastereoselective Additions of Oxindoles to Nitroolefins:Application to the Formal Synthesis of (+)-Physos- tigmine, J. Am. Chem. Soc. 131, 8758-8759, 2009.
Hernández-Rodríguez, M. y Juaristi, E., Structurally simple chiral thioureas as chiral solvating agents in the enantiodis- crimination of carboxylic acids, Tetrahedron 63, 7673- 7678, 2007.
Juaristi, E. Premio Nobel de Química 2001: La importancia de la síntesis asimétrica, Educ. quím. 13, 6-7, 2002. .
Juaristi, E. Síntesis asimétrica de aminoácidos valiosos, en Apor- taciones científicas y humanísticas mexicanas en el siglo XX, Paredes, O. y Estrada, S., Eds., Fondo de Cultura Económi- ca: México, 2008, p. 440-446.
Juaristi, E. Enantioselective synthesis of â-amino Acids, Wiley- VCH: Nueva York, 1997.
Juaristi, E. y Soloshonok, V. A. (Eds.) Second edition of enantiose- lective synthesis of â-amino acids, Wiley: Nueva York, 2005. Liu, Y., Melgar, R. y Juaristi, E. Enantioselective amination of
á-phenyl á-cyanoacetate catalyzed by chiral amines incor- porating the á-phenylethyl auxiliary, J. Org. Chem. 72, 1522-1525, 2007.
MacMillan, D. W. C., The advent and development of organo- catalysis, Nature455, 304-308, 2008.
Marigo, M., Juhl, K. y Jorgensen, K. A., Catalytic, highly enan- tioselective, direct amination of beta-ketoesters, Angew. Chem., Int. Ed. 42, 1367-1369, 2003.
Mukherjee, S.; Yang, J.W.; Hoffmann, S.; List, B., Asymmetric enamine catalysis, Chem. Rev. 107, 5471-5569, 2007.
Olivares-Romero, J. L. y Juaristi, E., Synthesis of Two Novel Chiral Diamines Derived from (S)-Proline and their Eva- luation as Precursors of Diazaborolidines for the Catalytic Borane-Mediated Enantioselective Reduction of Prochiral Ketones, Tetrahedron 64, 9992-9998, 2008.
Seayad, J. y List, B., Asymmetric organocatalysis, Org. Biomol. Chem. 3, 719- 724, 2005.
Simon,L.yGoodman,J.L.,MechanismofBINOL−Phosphoric Acid-Catalyzed Strecker Reaction of Benzyl Imines, J. Am. Chem. Soc. 131, 4070-4077, 2009.
Tanaka, K., Mori, A. y Inoue, S., The cyclic dipeptide cyclo[(S)- phenylalanyl-(S)-histidyl] as a catalyst for asymmetric addi- tion of hydrogen cyanide to aldehydes, J. Org. Chem. 55, 181-185, 1990.
- Detalles
- Alejandro Savin.
- Visto: 38898
Resonancia magnética nuclear
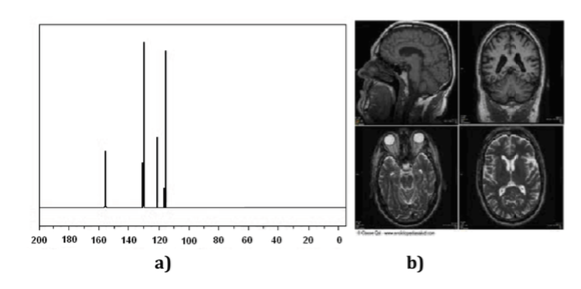
Las técnicas de Resonancia Magnética Nuclear (RMN) son un instrumento indispensable para la química así como para otras ramas de la Ciencia. Con la espectroscopia de RMN se pueden identificar moléculas (espectro Figura a), determinar su estructura o estudiar procesos dinámicos. Por ejemplo, ha sido clave en la determinación de la estructura de proteínas en disolución y, por otro lado, las técnicas de imagen de RMN son una herramienta indispensable en el diagnostico en medicina, como se ilustra en la (Figura b).
La RMN se sustenta en tres elementos:
a) El carácter magnético de los núcleos de las moléculas en estudio:
Muchos núcleos atómicos, debido al espín nuclear, presentan características magnéticas. En cierta manera se puede considerar que los núcleos se comportan como pequeños imanes.
b)La aplicación de un campo magnético intenso:
Cuando las moléculas en estudio están inmersas en un campo magnético, los niveles nucleares se desdoblan en varios niveles de energía. Cada uno de los niveles de energía que aparecen corresponden a diferentes orientaciones de los espines de los núcleos (pequeños imanes) respecto al campo magnético.
c) La iluminación de la muestra con radiación electromagnética:
Como en otras técnicas espectroscópicas la iluminación de la muestra con la frecuencia adecuada de radiación hará que los núcleos pasen de un nivel a otro. La frecuencia de la radiación necesaria para producir este salto de nivel, dependerá del tipo de núcleo, del entorno químico de éste, del tipo de núcleos presentes en sus cercanías y del campo externo aplicado. A lo largo de este tema se estudiarán los fundamentos físicos de esta técnica, y se explicarán las técnicas experimentales que permiten obtener los espectros, analizando, asimismo, las causas que hacen diferentes los espectros de las diversas moléculas.
Conceptos basicos de RMN:
Espín nuclear y momento angular de espín nuclear
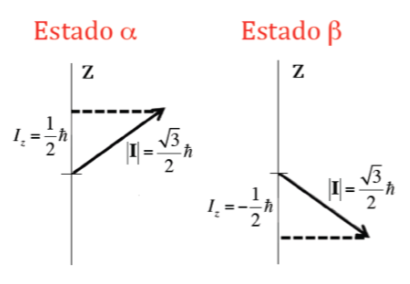
Los núcleos atómicos están formados por protones y neutrones. Ambos nucleones tienen momento angular orbital y momento angular de espín (tanto protones como neutrones tienen espín 1/2). La resultante de la suma vectorial de todos los momentos (orbitales y de espín) de todos los protones y neutrones del núcleo da como resultado el momento angular de espín nuclear y se simboliza por I y sus unidades son J s. Según la mecánica cuántica, el módulo del momento angular de espín nuclear, |I|, está cuantizado, con un valor I(I+1)hdonde I es el número cuántico de espín nuclear o espín nuclear y ħ es h/2p(que en el sistema internacional vale 1,05457·10‐34J·s). El valor de I es una característica fija de cada tipo de núcleo atómico y puede tomar unvalor entero o semientero (0,1, 2, 3,… ½ , 3/2 , 5/2….).
Además del módulo, también es importante conocer la componente del momento angular sobre el eje z, Iz. Esta puede tomar, asimismo, valores cuantizados dados por la expresión mIh donde mI es el número cuántico asociado a la componente z del momento angular de espín nuclear y que puede tomar los siguientes valores: –I, (‐I+1), ... , (I‐1), I. Así para un núcleo determinado, con un valor de I, existen (2I+1) orientaciones del momento angular de espín nuclear. Por ejemplo, considérese el núcleo atómico del isótopo más común del átomo de hidrogeno, 1H, que está formado por un protón y se sabe que su espín nuclear, I, es 1/2, y por tanto mI podrá tomar dos valores +1/2 y ‐1/2 (en RMN se suele denominar al isótopo 1H como protón). El protón tiene pues dos posibles estados nucleares uno denominado aconmI=+1/2 y otro bcon mI=‐1/2. En la Figura de mas adelante se muestran las dos posibles orientaciones del momento angular de espín nuclear correspondientes a los estados ayb, que energéticamente son equivalentes.
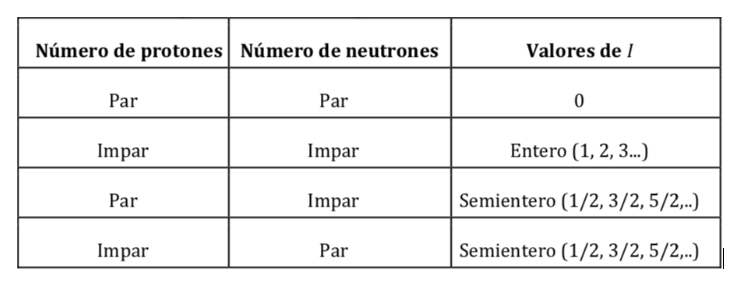
Valores de I según el número de protones y de neutrones del núcleo :
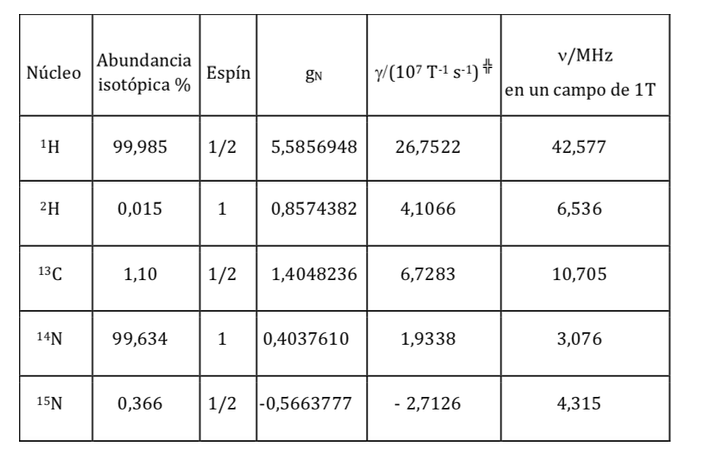
Propiedades magnéticas de diversos núcleos:
Momento dipolar magnético nuclear (magnetón nuclear, relación giromagnética ):
Así como el electrón tiene un momento dipolar magnético, algunos núcleos atómicos, pero no todos, presentan un momento dipolar magnético. Como se ha indicado, los núcleos se comportan como pequeños imanes, siendo el origen de este carácter magnético el momento angular del espín nuclear. El momento angular de espín nuclear, I, origina un momento dipolar magnético nuclear mI que viene dado por:
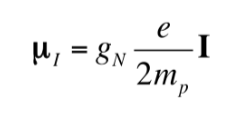
donde gN es el factor g nuclear. Obsérvese que en esta expresión se utiliza por definición la masa y carga del protón (mp y e), en lugar de la masa y la carga del núcleo en cuestión. Las diferencias de un núcleo a otro se engloban en el factor gN nuclear. El valor de gN no puede obtenerse teóricamente, por lo que se determina experimentalmente. Las unidades de mI son J T‐1
La relación entre mI eI, también se puede expresar de la siguiente forma:
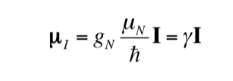
con
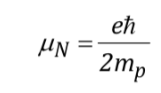
siendo una constante con un valor de 5,050 10‐27J T‐1 denominadamagnetón nuclear y ges la relación giromagnética (T‐1s‐1).En las tablas anteriores ya se mostraron valores de estas magnitudes para algunos isótopos. Aunque en la mayoría de los casos mI e I tienen el mismo sentido, a veces es opuesto, como se deduce de los valores negativos del factor gN y de gpara el isótopo 15N, por ejemplo.
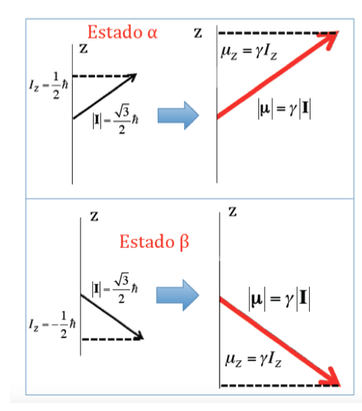
En el caso del protón donde se habían visto dos posibles orientaciones del momento angular se tendrán dos posibles orientaciones del momento magnético dipolar, como se ilustra en la siguiente figura , siendo ambas orientaciones energéticamente equivalentes, ya que la elección de una dirección z es arbitraria.
Energía de interacción del campo magnético con el momento magnético nuclear:
Si se introduce una molécula dentro de un campo magnético, con densidad de flujo o campo magnético B, el momento magnético dipolar de cada uno de sus núcleos interaccionará con él, como se ilustra en la siguiente figura , viniendo expresada la energía de la interacción por:
![]()
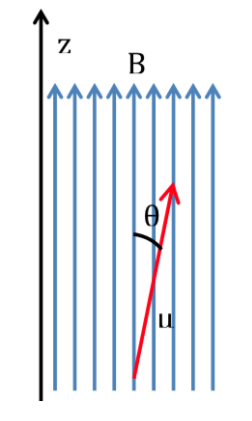
Según la física clásica, todas las orientaciones de un dipolo magnético dentro de un campo magnético son permitidas y por tanto qpuede tomar cualquier valor entre 0 y 180o. Dependiendo de la orientación del dipolo la energía será mayor o menor:
La interacción más favorable se da cuando ambas magnitudes tienen el mismo sentido: q=0 y por tanto cos q=1 y :
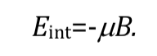
Sin embargo, según la mecánica cuántica, no todas las orientaciones del dipolo magnético nuclear están permitidas, las orientaciones están cuantizadas y sólo son posibles unos determinados valores de q. Hay tantas orientaciones permitidas como valores de Iz o de mI (‐I, ‐I+1,..., I‐1, I) es decir 2I+1 orientaciones. Evidentemente el número de orientaciones del momento dipolar varía con el tipo de núcleo atómico. Para poder definir las orientaciones se toma la dirección y sentido del campo B como referencia del eje Z.
Así la energía de interacción del campo magnético exterior B y el dipolo nuclear vendrá dada por: j
Frecuencia de Langmor
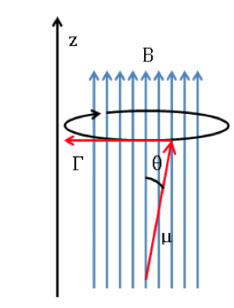
Si sobre el núcleo actúa un campo magnético externo de intensidad B, el dipolo magnético nuclear experimenta un par de fuerzas como se muestra en la siguiente formula dado por:
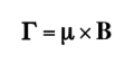
que tiende a alinear el dipolo con el campo magnético. Como el dipolo magnético no puede alinearse con el campo magnético, el sistema no es capaz de disipar esta energía y describe un movimiento de precesión, denominado precesión de Larmor, en
torno a la dirección del campo aplicado. La frecuencia de esta precesión se llama frecuencia de Larmor, con un valor:
Esquema de langmor :
Espectroscopia de RMN
La clave de cualquier técnica espectroscópica se basa en la posibilidad de que la molécula en estudio pueda estar en más de un nivel de energía, de tal forma, que haciendo incidir radiación electromagnética (r.e.m) de la frecuencia adecuada, se pueda pasar a nuestro sistema (molécula) de un nivel energético a otro. Como se ha visto en el apartado anterior, el núcleo de un protón, inmerso en un campo magnético, puede ocupar dos niveles diferentes asociados al espín nuclear, por tanto haciendo incidir un haz de r.e.m de la frecuencia adecuada sobre un núcleo de 1H que esté ocupando el nivel inferior, se le puede pasar al nivel superior. Este tipo de transiciones es lo que da lugar a la Espectroscopia de Resonancia Magnética Nuclear (RMN). A partir de ahora nos centraremos en el caso del protón 1H, por ser la espectroscopia más empleada, aunque todo lo que se indique es aplicable a otros núcleos. De hecho, cada vez se utiliza más variedad de núcleos.
Frecuencia de resonancia:
La frecuencia que provoca la transición se obtiene aplicando la condición de resonancia:
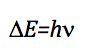
Como la diferencia de energía entre dos niveles consecutivos es:
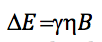
igualando ambas expresiones y despejando n:
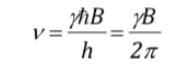
que, como se puede observar, coincide con la frecuencia de precesión de Larmor.
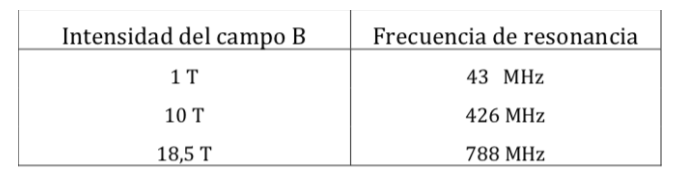
Frecuencias de resonancia de RMN de protón para diversas intensidades de campo :
Reglas de selección :
Las reglas de selección que se estudiaron en otras espectroscopias corresponden a la interacción del dipolo eléctrico con el campo eléctrico de la radiación. En cambio en RMN la interacción se produce entre el dipolo magnético del núcleo y el campo magnético variable de la radiación. Para determinar las reglas de selección, se debe recordar la expresión general que determina la probabilidad de paso entre dos estados es:
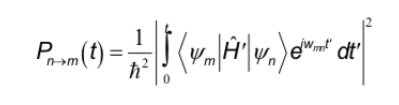
El hamiltoniano, en este caso, es el producto del operador del campo magnético oscilante de la radiación por el operador del momento dipolar del núcleo.
![]()
Se puede demostrar que para que haya una transición entre dos niveles la correspondiente regla de selección es:
mI=±1
Intensidad de la señal (población de los niveles ):
La intensidad de la señal está directamente relacionada con la población de los niveles involucrados en la transición. Según la ley de distribución de Boltzman, para un sistema de dos niveles, como el protón, el cociente de la población del nivel superior, Nb, y la población del nivel inferior Na, viene dado por:
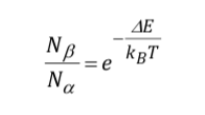
Descripción de los equipos de RMN :
¿Qué procedimiento experimental se utiliza par obtener el espectro? Generalmente en espectroscopia se varía la frecuencia de la radiación incidente, hasta que se observa absorción. Sin embargo, en espectroscopia RMN se tiene además la opción de mantener fija la frecuencia de la radiación y variar el espaciado entre los niveles, variando la magnitud del campo aplicado B hasta que se observe absorción. En la siguiente figura donde se presenta un esquema de un equipo de RMN. La muestra se coloca en un tubo, que, algunas veces, se gira rápidamente para ganar homogeneización en la señal, aunque actualmente está en discusión el uso del giro, pues puede introducir errores. El tubo, a su vez, está situado entre dos polos magnéticos que son los que generan el campo magnético. Hay un emisor y un receptor de radiofrecuencia. La señal del emisor puede ser controlada para hacer un barrido de frecuencias. Con las bobinas de barrido se puede controlar un barrido del campo. Los equipos actuales de RMN ya casi no emplean :
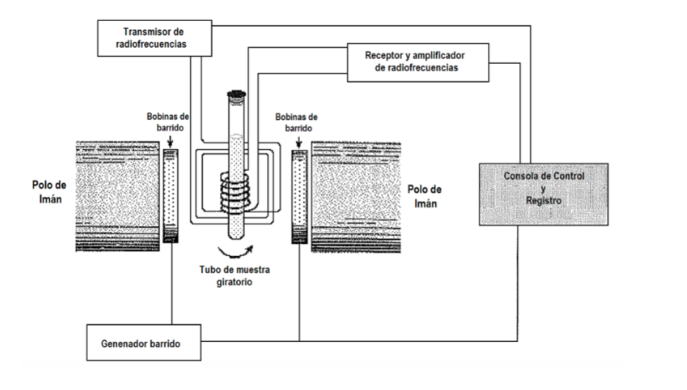
los procedimientos anteriores, sino que se basan en la utilización de técnicas de transformadas de Fourier. B se mantiene fijo y la muestra se ilumina con un pulso de radiofrecuencias de duración determinada y de alta potencia con frecuencia nrad, un valor fijo en el intervalo de frecuencias de RMN del tipo de núcleo que se esté estudiando. El pulso dura varios microsegundos, debido a lo cual, se puede demostrar matemáticamente por una técnica llamada análisis de Fourier, que el pulso de radiofrecuencias es equivalente a un intervalo de frecuencias ∆nen torno a nrad y excita todas las posibles resonancias de RMN. La señal observada en el detector contendrá todas las frecuencias de resonancia y se analiza con un ordenador utilizando técnicas de transformada de Fourier. Este procedimiento permite muy alta resolución y trabajar con núcleos que con los procedimientos anteriores tenían señales muy débiles.
Apantallamiento nuclear:
En resonancia Magnética se utilizan núcleos con momento angular de espín distinto de cero, como pueden ser 1H y 13C. Sin embargo, las frecuencias de resonancia no son iguales para todos los núcleos de hidrógeno o de carbono, dependen del entorno químico que rodea cada núcleo. Esto se debe a que los electrones que rodean cada núcleo generan un campo magnético que se opone al aplicado, se dice que los núcleos están apantallados, siendo σ la constante de apantallamiento.
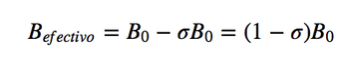
Bef es el campo magnético neto que actúa sobre el protón; B0 es el campo magnético aplicado; σ es la constante de apantallamiento, independiente del campo aplicado. Bajo esta nueva situación, con los núcleos apantallados por la densidad electrónica que los rodea, la frecuencia de resonancia pasa a ser
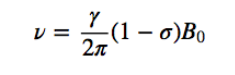
Los núcleos con distinto entorno químico presentan una constante de apantallamiento diferente, generando diferentes señales en el espectro de RMN.
Espectro del etanol:
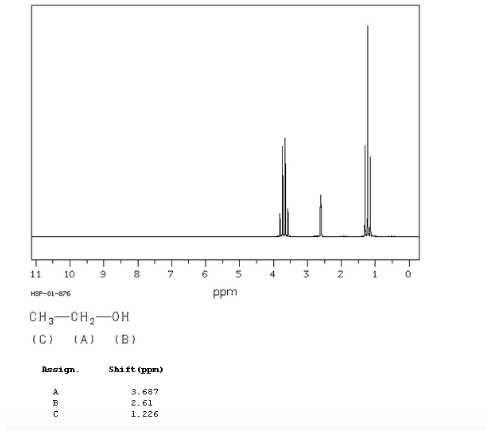
En el espectro de RMN del etanol se observan tres señales diferentes, debido a la existencia de 3 tipos de hidrógenos con distinto entorno químico. Los hidrógenos A están más desapantallados que los C debido a la presencia del oxígeno (átomo electonegativo que retira densidad electrónica). El entorno químico del hidrógeno B, unido directamente al oxígeno, también es diferente resonando a una frecuencia distinta a los anteriores.
Desplazamiento quimico
Las señales del espectro de RMN se miden en una escala independiente del campo magnético aplicado, llamada desplazamiento químico y representada por la letra δ. Independientemente del campo magnético al que trabaje el espectrofotómetro, las señales de un compuesto químico se obtienen siempre a los mismos valores de δ:

Tabla de desplazamientos:
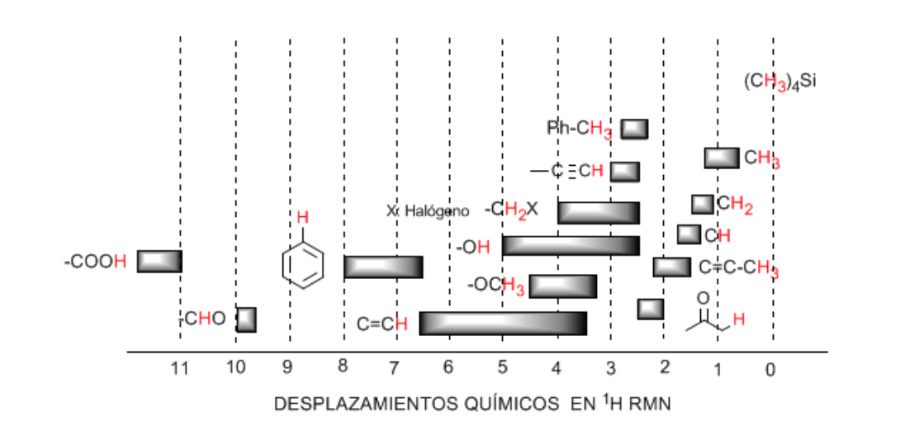
Los hidrógenos situados sobre cadenas alifáticas presentan valores de δ próximos a 1. Aunmentando ligeramente al pasar de carbonos primarios a secundarios o terciarios.
Los hidrógenos alílicos se localizan entre 1,5 y 2,1.
Los hidrógenos alfa respecto a carbonilos y derivados de ácido se sitúan entre 2 y 2,5.
Los hidrógenos bencílicos entre 2,3 y 2,7.
El hidrógeno de alquinos terminales entre 2,5 y 3.
Hidrógenos unidos a carbonos con halógenos entre 2,5 y 4 dependiendo de la electronegatividad del halógeno
Los hidrógenos del grupo hidroxilo entre 2,5 y 5. Rango muy amplio debido a la formación de puentes de hidrógeno.
Hidrógenos de carbonos unidos a oxígeno tipo éter entre 3,3 y 4,5.
Hidrógenos olefínicos entre 3,5 y 6,5.
Hidrógenos unidos a sistemas aromáticos entre 6,5 y 8.
Hidrógeno de aldehídos 9,5-10
Hidrógeno del grupo ácido carboxílico por encima de 11.
Los grupos electronegativos desapantallan los nucleos :
Los sustituyentes electronegativos retiran densidad electrónica, desapantallando los hidrógenos y desplazan la señal hacia valores altos de δ.
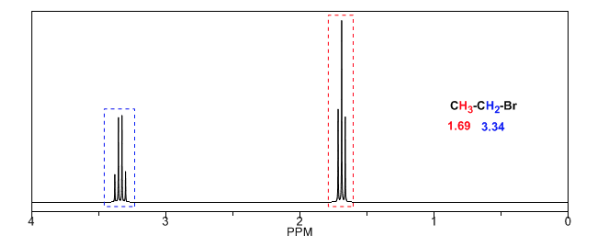
En la siguiente tabla puede verse la influencia de diferentes átomos sobre la señal de los hidrógenos del metilo.
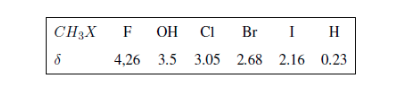
En los siguientes espectros puede observarse los efectos comentados anteriormente sobre los desplazamientos químicos.
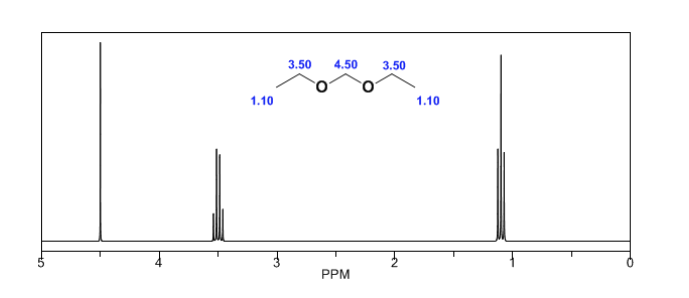
Anisotropia magnetica:
Los protones próximos a dobles enlaces y anillos aromáticos están especialmente desapantallados debido al campo magnético inducido por las corrientes electrónicas de estos sistemas. El campo inducido se suma al aplicado, produciendo un desplazamiento superior al esperado.
En la siguiente imagen podemos ver la cirulación electrónica (curvas en negrita) y el campo magnético inducido (lineas a trazos) para un alqueno y un carbonilo. Obsérvese como en la región del protón el campo magnético inducido tiene identica dirección y sentido que el aplicado.
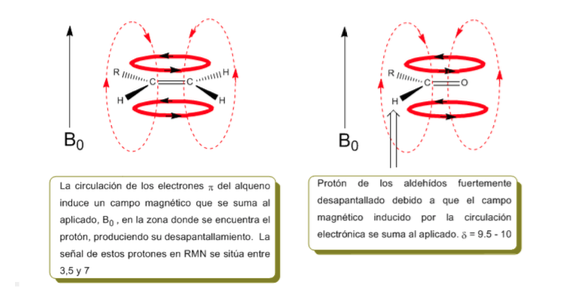
En el caso del benceno se observa una situación análoga. Sin embargo, en los alquinos la circulación electrónica induce un campo magnético que se opone al aplicado en la zona del protón. Los hidrógenos acetilénicos están apantallados con señales en el espectro de RMN a desplazamientos bajos.
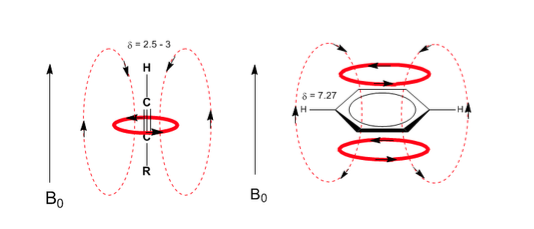
Obsérvese como los campos inducidos aumentan considerablemente los deplazamientos del protón olefínico, viéndose también afectadas las posiciones alílicas.
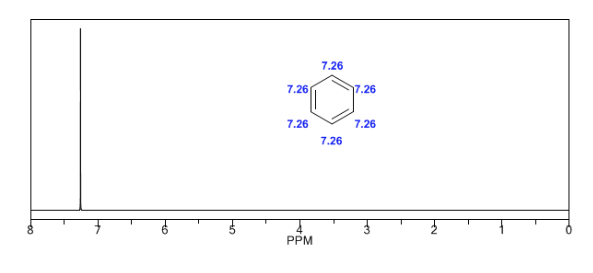
Los hidrógenos aromáticos están fuertemente desapantallados debido al campo inducido por las corrientes del anillo.
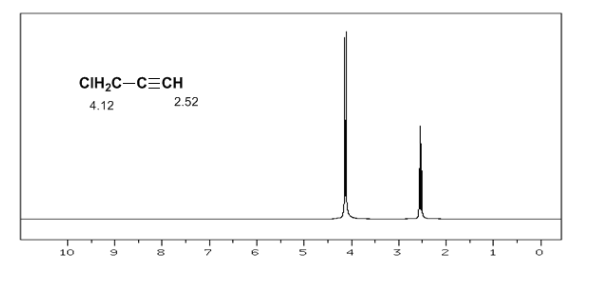
El hidrógeno acetilénico presenta un desplazamiento bajo, debido a que las corrientes producen un campo magnético que se opone al aplicado.
Los desplazamientos hidrógenos ácidos más comunes en las moléculas orgánicas son:
- ·Ácidos carboxílicos (RCOOH) δ= 10 - 12 ppm
- ·Aminas (R−NH2) δ= 0.5 - 5 ppm
- ·Amidas (RCONH2) δ= 5-8 ppm
- ·Alcoholes (ROH) δ= 0.5 - 5 ppm
- ·Fenoles (Ph-OH) δ= 4 - 7 ppm
Acoplamiento espin-espin:
La información estructural del RMN deriva de dos factores: los diferentes desplazamientos observados dependiendo del ambiente químico que rodea al protón y del acoplamiento entre los espines de protones próximos, que produce el desdoblamiento de las señales.
Aunque algunas señales del espectro son picos simples, es habitual encontrar señales compuestas por varios picos muy próximos, que se nombran con la siguiente notación: singulete (s), doblete (d), triplete (t), cuatriplete (c), quintuplete (q), sextuplete (sx) y septuplete (sp), señales complejas se las designa como multipletes. El valor de δ de estas señales se asigna al centro de las mismas, salvo que el multiplete sea irregular en cuyo caso se indica el intervalo.
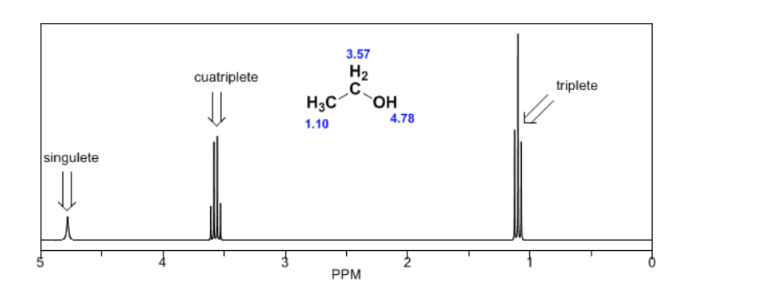
En el espectro del etanol puede observarse que el hidrógeno hidroxílico produce un singulete, la pareja de hidrógenos del carbono uno dan lugar a un cuatriplete y los tres hidrógenos del carbono dos producen un triplete.
Explicación del acoplamiento espín-espín.
Para comprender el desdoblamiento de las señales debido al acoplamiento espín-espín vamos a estudiar el espectro del 1,1-dicloro-2,2-difeniletano (Cl2CHaCHbPh2).
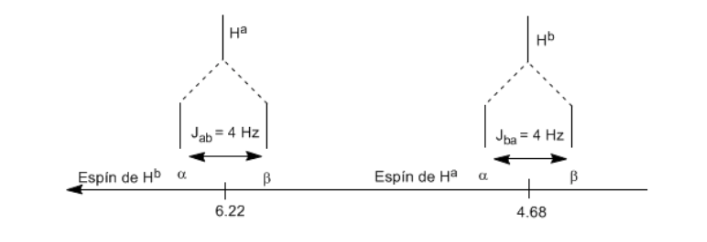
El proton Ha sometido a un campo magnético B0 produce una señal a δa=6,22ppm. Sin embargo, el protón Hb genera un pequeño campo magnético que afecta al protón Ha. Aproximadamente la mitad de las moléculas tienen el protón Hb alineado con el campo aplicado (espín alfa) y la otra mitad lo tienen orientado en contra del campo (espín beta). Cuando Hb tiene espín α, Ha se ve sometido a un campo ligeramente mayor y resuena a una mayor frecuencia (δ ligeramente mayor). Cuando Hb tiene espín β, Ha se ve sometido a un campo ligeramente menor y resuena a menor frecuencia (δ ligéramente menor), lo cual produce el desdoblemiento del pico inicial en dos señales separados por una distancia de 4 Hz, llamada constante de acoplamiento (J). Este mismo razonamiento se puede realizar para el proton Hb.
Por último, discutiremos el acoplamiento de un protón con tres protones vecinos equivalentes. En este caso se observa una señal formada por cuatro picos (cuatriplete). Los picos centrales poseen el triple de intensidad que los picos de los extremos.
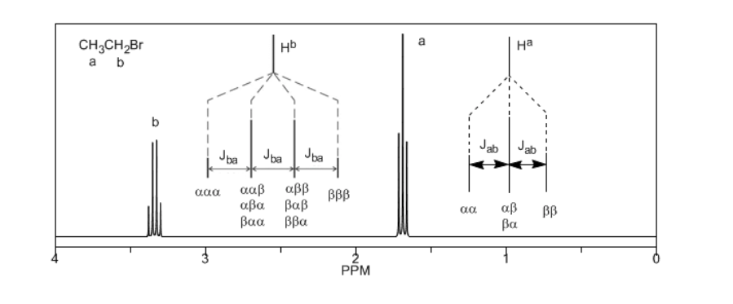
Regla N+1
De la discusión anterior puede deducirse que un protón produce una señal con un número de picos una unidad superior al número de hidrógenos vecinos. En la siguiente imagen podemos observar los picos que produce un hidrógeno Hb al acoplarse con un número de hidrógenos variables
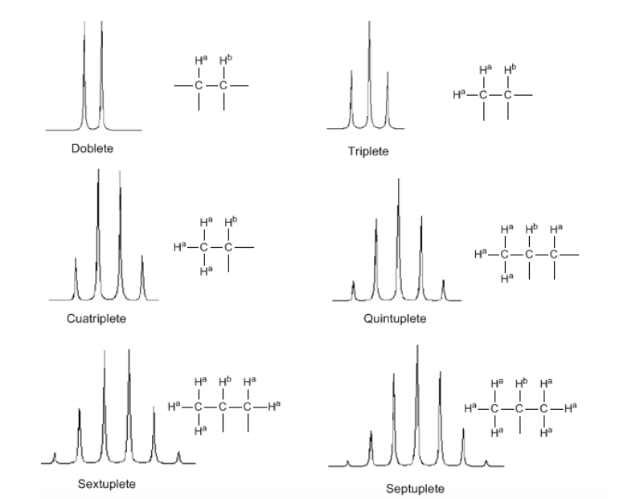
La intensidad de los picos de una señal vienen dados por el triángulo de Pascal (Tartaglia):
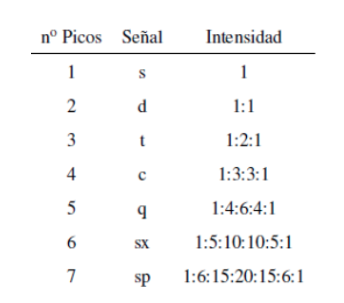
Deben tenerse en cuenta dos consideraciones al aplicar la regla N+1:
- En moléculas del tipo A−CHa2−CHb2−CHa2−Alos protones Hb aparecen como un quintuplete.
- En moléculas del tipo A−CH2−CH2−A, los cuatro protones son equivalentes y dan un singulete.
Acoplamiento de tres nucleos no equivalentes:
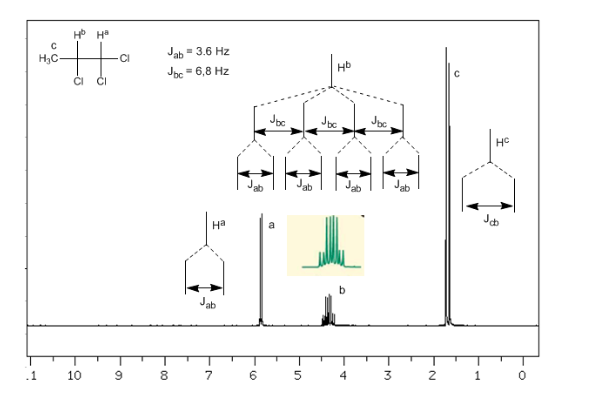
Teniendo en cuenta que el orden de constantes de acoplamiento en alquenos es Jtrans>Jcis>Jgeminal, los árboles de acoplamiento para los hidrógenos Ha y HM son los indicados en el espectro. ¿Serías capaz de dibujar el arbol para Hx?
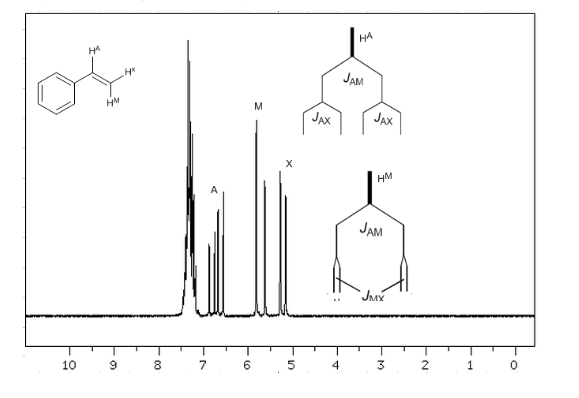
Resonancia magnética nuclear de 13C
La resonancia magnética nuclear de 13C es complementaria a la de 1H. Esta última técnica se utiliza para deducir la estructura del esqueleto carbonado observando los entornos magnéticos de los átomos de hidrógeno, mientras que la espectroscopia de RMN de 13C determina el entorno magnético de los átomos de carbono.
Aproximadamente el 99% de los átomos de carbono en una muestra natural son del isótopo 12C. Este isótopo posee un número par de protones y un número par de neutrones, por tanto, no tiene espín magnético y no puede dar lugar a señales de resonancia magnética nuclear. El isótopo de 13C menos abundante tiene un número impar de neutrones, lo que le confiere un espín magnético de 172, igual al del protón. La espectroscopia de resonancia magnética nuclear de 13C es menos sensible que la de 1H debido a que sólo el 1% de los átomos de carbono posee espín y a que, además, la frecuencia de resonancia del 13C, para un campo magnético dado, es la cuarta parte de la que se da en la RMN de 1H.
Los desplazamientos químicos del carbono son de 15 a 20 veces mayores que los del hidrógeno debido a que el carbono está directamente unido a los átomos que resultan ser bien apantallantes o desapantallantes. Por ejemplo, el protón de un aldehído absorbe a 9.4 ppm en el espectro de 1H mientras que el carbono de carbonilo absorbe a 180 ppm en el espectro de 13C.
Además, las señales en el espectro de 13C son líneas verticales, es decir, no hay desdoblamientos de espín-espín. Esto se debe a que sólo el 1% de los átomos de carbono entran en resonancia, y por tanto, existe una probabilidad muy pequeña de que un núcleo de 13C esté adyacente a otro núcleo de 13C.
A continuación se da una tabla de valores aproximados de desplazamientos químicos en un espectro de resonancia magnética nuclear de 13C:
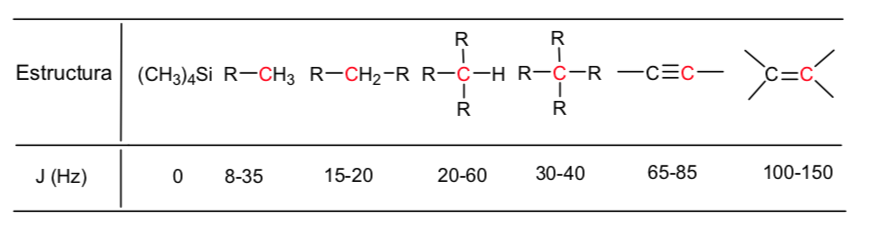
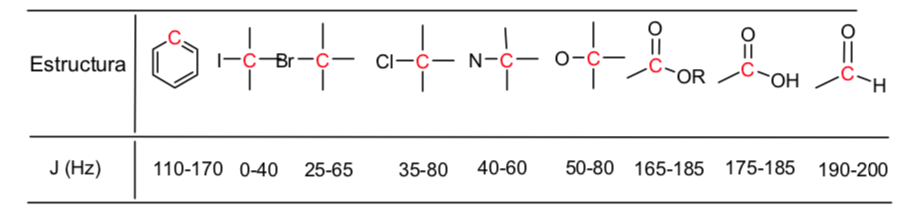
Referencias recomendadas para expandir la busqueda :
1. Aga Fano S.A. Espectroscopia de Emisión. (acceso 10 de septiembre del 2007).http://hiq.aga.com.co/International/Web/LG/CO/likelgspgco.nsf/DocByAlias/anal_icp.
2. Alonso, P. et al. quimicaCou..Ed. Mc Graw-Hill. 1990.
3. Álvarez Jiménez, M. D. y Gómez del Río, M. I. Guía didacticaQuímica Analítica II. UNED. 1999.
4. Arribas Jimeno Siro; Burriel Barcelo Fernando; Hernández Méndez Jesús; Lucena Conde Felipe. Química Analítica Cualitativa. ISBN: 8497321405. ISB. 2006.
5. Ayres, Gilbert H. Análisis Químico Cuantitativo. Ediciones del Castillo, 4ta ed. ISBN: 8421902806. 1981.
6. Bermejo Barrera. M del Pilar. Química analítica general, cuantitativa e instrumental. Editorial Paraninfo. 7ma Edición. ISBN: 8428318093. 1990.
7. Blanco, M., Cerdá, V. y Sanz Medel, A., Espectroscopia Atómica Analítica, Publicaciones de la universidadAutónoma de Barcelona. 1990.
8. Brode. R.W, Chemical spectroscopy, Nueva York 1952.
9. Burriel, M.F., Lucena, C.F.Química Analítica Cuantitativa. Edición Revolucionaria. La Habana.1978.
10. Burriel, F. Química Analítica Cualitativa. Editorial Paraninfo. ISBN: 8497321405. pp 1072. , 2003.