Estereoquímica
Estereoquímica é o estudo de compostos orgânicos no espaço. Para compreender as propriedades dos compostos orgânicos é necessário considerar todas as três dimensões espaciais. As bases da estereoquímica foram lançadas por Jacobus van't Hoff e Le Bel, no ano de 1874, bem como por Ernest L. Eliel no século XX. Eles propuseram independentemente que os quatro substituintes em um ponto de carbono para os vértices de um tetraedro, com o carbono no centro do tetraedro.
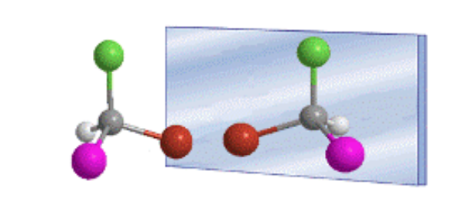
O arranjo tetraédrico dos substituintes em um carbono sp 3 dá origem à existência de dois compostos possíveis, que são imagens especulares não sobreponíveis, chamados enantiômeros.
Em geral, moléculas que diferem pelo arranjo espacial de seus átomos são chamadas de estereoisômeros.
Isomeria
Isômeros são aqueles compostos que possuem fórmulas moleculares idênticas, mas que diferem na natureza ou arranjo das ligações entre seus átomos ou no arranjo de seus átomos no espaço.
Para interpretar as diferenças nas propriedades, os químicos do século passado imaginaram que os átomos de uma molécula tinham arranjos espaciais particulares que explicavam seus diferentes comportamentos.
A classificação por função química, estabelecida de acordo com o comportamento dos compostos, tem sido relacionada à presença na molécula de um grupo de átomos denominado grupo funcional.
Além da importância do grupo funcional, há uma diferença de comportamento induzida por pequenas diferenças no arranjo dos diferentes átomos que compõem o restante da molécula. Essas diferenças podem responder a diferentes classes de isomeria:
função isomerismo
Os isômeros constitucionais, que diferem entre si porque seus grupos funcionais são diferentes, pertencem a esse tipo de isomeria.
O grupo funcional em ambos os isômeros é diferente
C2H6O_ _
Etanol (CH 3 -CH 2 -OH) e éter dimetílico (CH 3 -O-CH 3 )
Álcool reage com sódio enquanto com éter nenhuma reação é observada.
Do ponto de vista físico, o álcool é um líquido com ponto de ebulição de 78,5°C, enquanto o éter é um gás que se liquefaz a -23°C.
Isomeria de posição e/ou esqueleto.
Os grupos funcionais são idênticos, mas estão colocados em posições diferentes no esqueleto molecular (isômeros posicionais).
Ex: 2-hexanol e 3-hexanol:
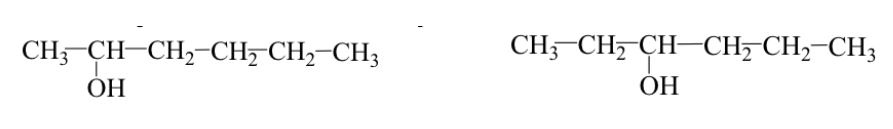
Às vezes, o grupo alquil tem um arranjo diferente (espinha dorsal ou isômeros ramificados).
Ex: 3-metil-2-pentanol e 2-hexanol
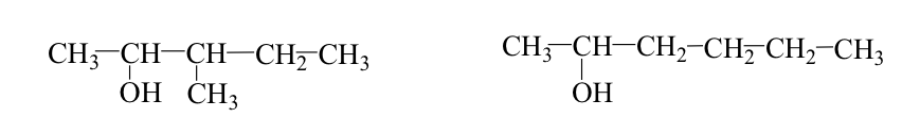
Ambos os casos podem ocorrer simultaneamente:
Ex: 3-metil-2-pentanol e 3-hexanol
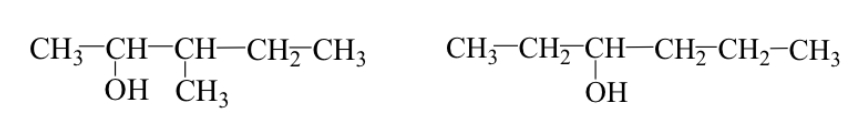
tautomerismo
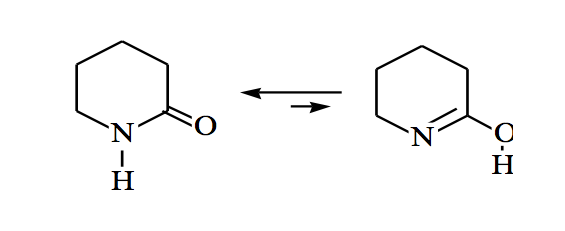
São isômeros constitucionais de fácil interconversão, pois estão em rápido equilíbrio entre si. O fenômeno é chamado de tautomerismo e geralmente consiste em um átomo, geralmente hidrogênio, localizado em uma tríade de átomos e uma ligação dupla mudando de posição simultaneamente.
O exemplo mais clássico é o equilíbrio ceto-enol ( -ene para a ligação dupla e -ol para o álcool).
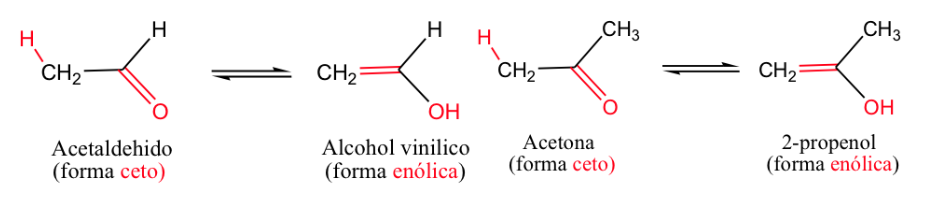
Geralmente, as formas de cetona são as mais estáveis, mas quando a forma de enol se estabiliza (por ligação de hidrogênio ou por ressonância) o equilíbrio muda.
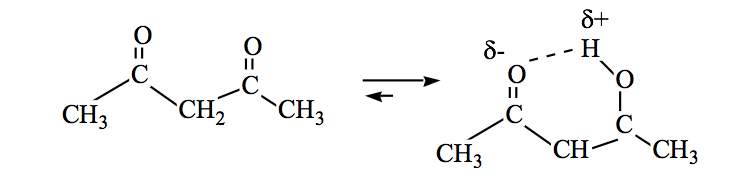
As amidas também podem estar em equilíbrio ceto-enol:
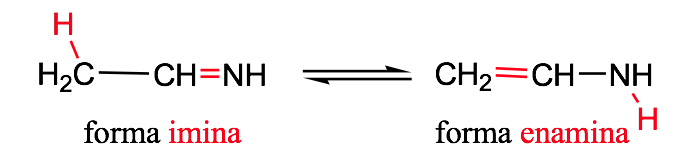
Tautomerismo de imina-enamina :
nitro-acido
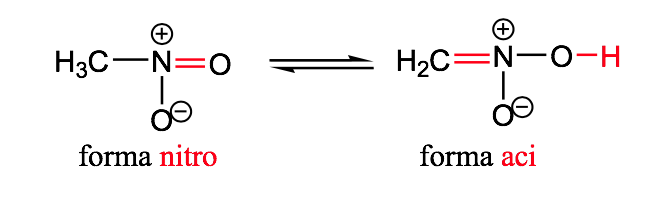
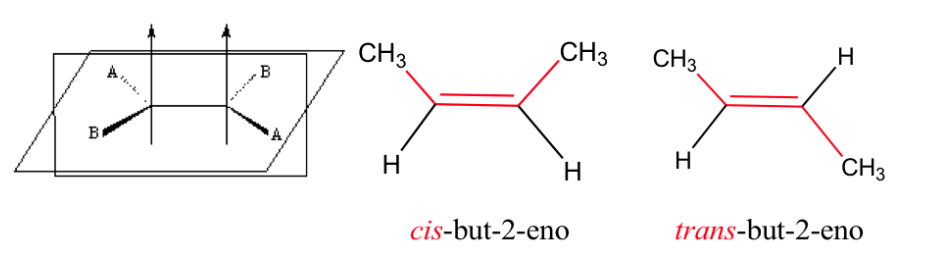
isomerismo geométrico
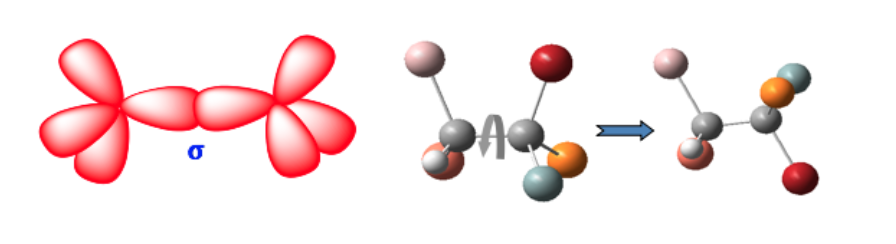
Essas duas formas não são isômeros geométricos, pois a rotação livre em torno da ligação simples converte uma forma em outra (conformes).
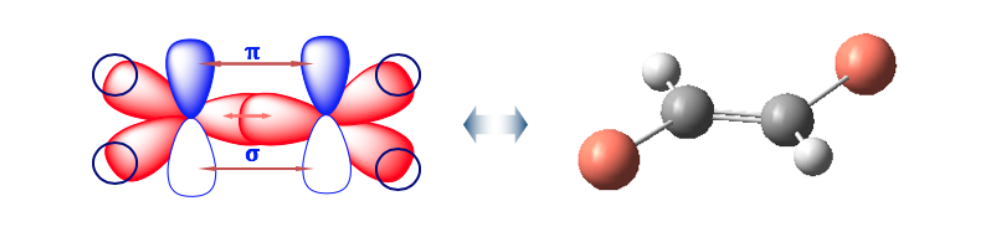
A ligação dupla não permite rotação livre, o que pode gerar duas estruturas diferentes dependendo da posição dos grupos A e B no espaço: são isômeros geométricos .
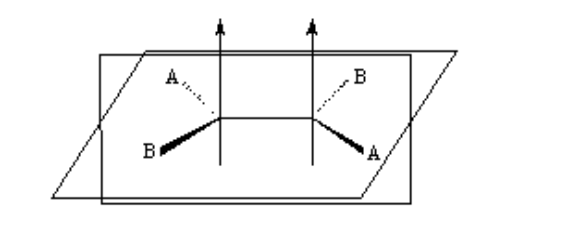
isomerismo em alcenos
Para que exista isomeria geométrica, duas condições devem ser atendidas:
1.- Rotação impedida (por exemplo por uma ligação dupla)
2.- Dois grupos diferentes (A e B) fixados em ambos os lados do link
Efeito do isomerismo geométrico nas propriedades físicas

Nomenclatura de isômeros geométricos
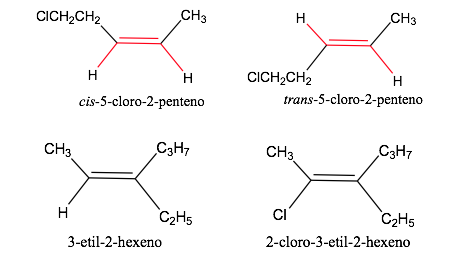 Regras de sequência ou prioridade
Regras de sequência ou prioridade
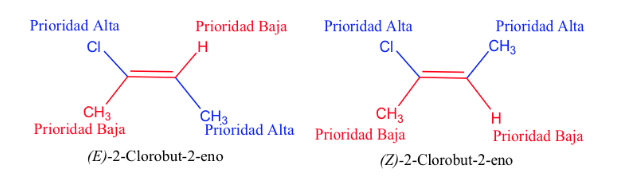
As regras que devem ser levadas em conta para estabelecer a ordem de prioridade ou preferência de átomos ou grupos de átomos foram estabelecidas em 1956 por Cahn , Ingold e Prelog e modificadas várias vezes para evitar ambiguidades.
Quando os grupos de alta prioridade estão em lados opostos do plano perpendicular à molécula, o isômero é chamado de E.
Quando estão do mesmo lado desse plano, o isômero é denominado Z.
Exemplo: but-2-eno:
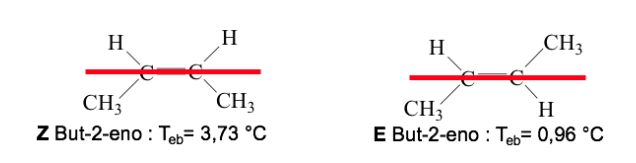
A prioridade dos substituintes nos carbonos da ligação dupla pode ser deduzida das seguintes regras:
- Regra 1:
- Se os átomos ligados ao átomo de carbono em estudo forem diferentes, aqueles com maior número atômico têm prioridade sobre aqueles com menor número atômico e se houver dois isótopos, eles são considerados em ordem decrescente de massa atômica.
Ex: Br (35)> Cl (17)> O (8)> N (7)> C (6)> H (1)
D> H e 13 C> 12 C
Regra 2 :
Quando os átomos ligados ao átomo de carbono são idênticos (e a primeira regra não funciona), segue-se a sequência, ou seja, comparam-se os átomos ligados a eles e, se necessário porque também eram iguais, comparam-se os átomos ligados a eles são usados, a seguir, etc., tendo em conta que se os átomos são iguais mas em número diferente, tem prioridade o substituinte com mais átomos de ordem superior.
Ex: -CH 2 -OH> -CH 3 porque O> H
-CH 2 -Br> -CH 2 -OH porque Br> OH
-CH 2 -CH 3> -CH 3 porque C> H
Regra 3:
As ligações duplas e triplas são tratadas como se fossem simples, duplicando ou triplicando os átomos da cadeia, respectivamente.
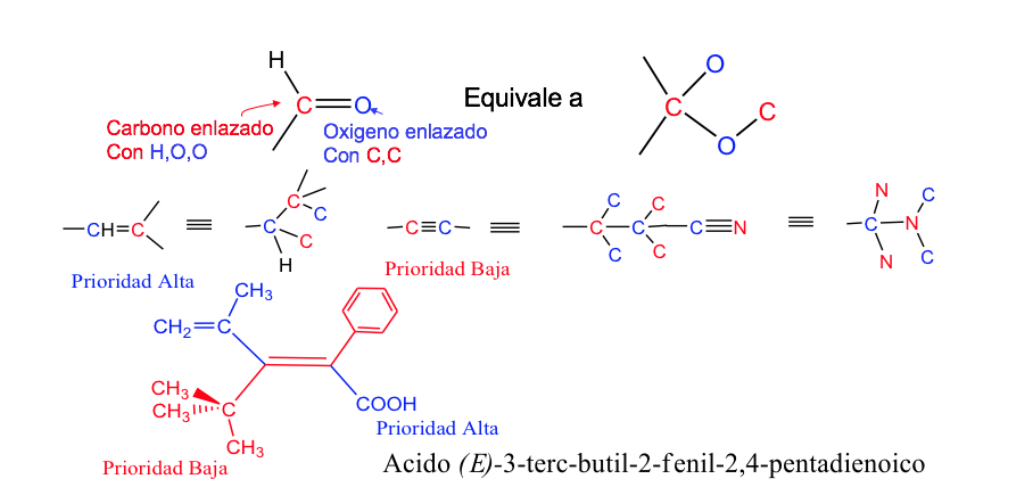
. Isomeria de ciclos e sistemas complexos.
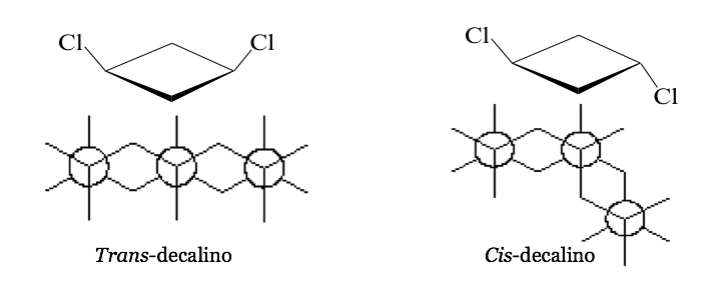
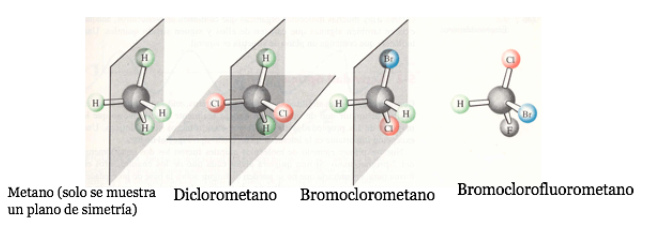
Em algumas moléculas cíclicas simétricas (pelo menos dissubstituídas), os átomos do ciclo definem um plano. Um substituinte fica em direção a uma face deste plano, enquanto o outro pode ficar no mesmo lado ou no lado oposto.
cis- 1,3-Diclorociclobutano trans- 1,3-Diclorociclobutano
Representação projetiva (CRAM)
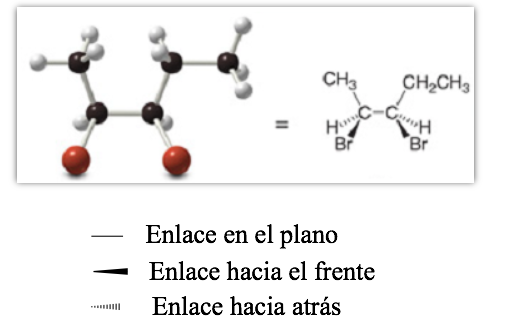
representação em perspectiva
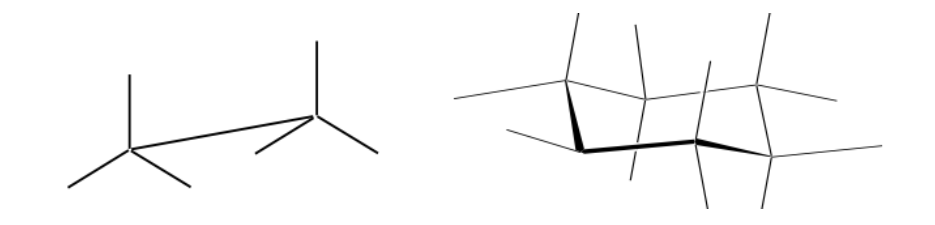
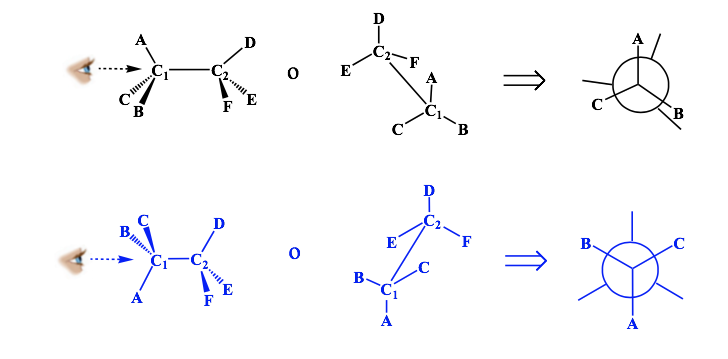
Representação de Newman
Uma projeção de Newman é uma forma de representação bidimensional útil para visualizar conformações em uma ligação simples carbono-carbono em uma molécula orgânica.
Representação de Fisher.
A projeção de Fisher é uma forma padrão de desenhar átomos de carbono tetraédricos e seus substituintes em duas dimensões.
Nesta projeção cada carbono tetraédrico é representado como uma cruz na qual as linhas horizontais são direcionadas para fora do papel e as verticais para dentro.
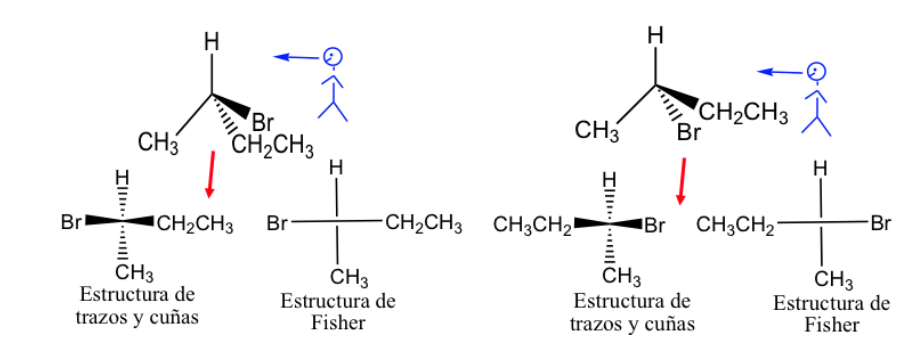
A representação de Fisher pode ser feita marcando as ligações que estão na frente do plano com uma linha grossa e as localizadas atrás com uma linha tracejada, mas geralmente as diferentes ligações são apresentadas com linhas normais, embora entenda-se que os substituintes representados a a direita e a esquerda da linha vertical estão acima do plano de representação e as representadas acima e abaixo estão abaixo desse plano.
Por convenção geral, a cadeia carbônica é apresentada verticalmente, colocando o carbono mais oxidado no topo.
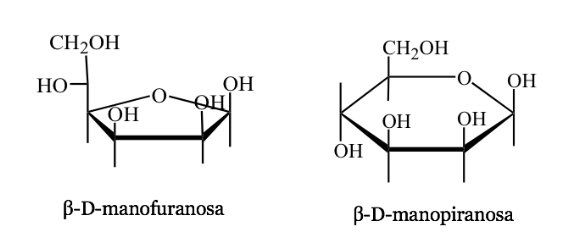
Representação de Harwoth
- É uma representação em perspectiva das formas cíclicas de moléculas de açúcar com 5 ou 6 átomos (furanoses, piranoses).
- Por exemplo:
quiralidade
Qualquer figura geométrica, ou qualquer grupo de pontos, cuja imagem em um espelho plano, idealmente realizada, não pode ser feita para coincidir com ela mesma, é chamada quiral. Algumas moléculas são como mãos. A esquerda é a imagem espelhada da direita, mas não são sobreponíveis e, portanto, não são idênticas. Eles são chamados quirais .
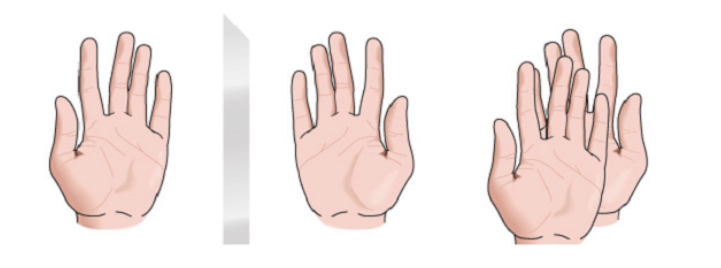
Existem outras moléculas semelhantes a um par de meias. As meias são imagens espelhadas umas das outras e também são sobreponíveis.
Uma molécula ou um objeto sobreponível em sua imagem no espelho é chamado aquiral .
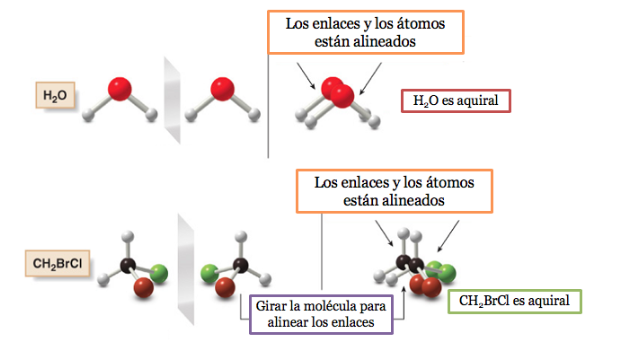
Se uma molécula tem um plano de simetria, é um sistema aquiral.
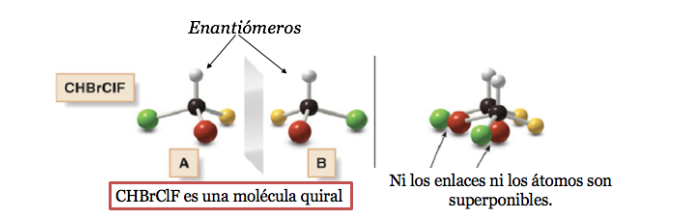
Uma molécula quiral existe em duas formas estereoisoméricas chamadas enantiômeros . Estes são objetos não sobreponíveis com suas imagens espelhadas.
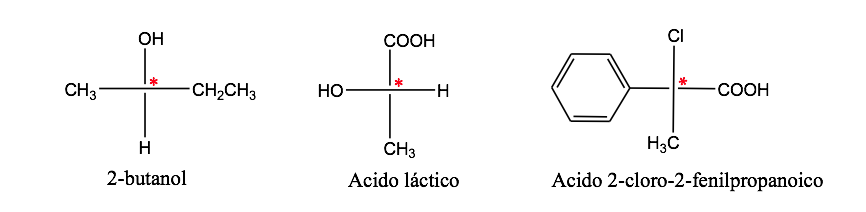
Um átomo de carbono ligado a 4 substituintes diferentes é chamado de carbono assimétrico . Entretanto, sua existência não é garantia de quiralidade (como veremos adiante). Também chamado de carbono estereogênico ou estereocentro.
isomerismo óptico
As propriedades físicas de dois enantiômeros são idênticas: eles têm os mesmos pontos de ebulição e fusão, a mesma solubilidade, a mesma densidade, o mesmo índice de refração, a mesma condutividade... etc.
A atividade óptica dos pares de enantiômeros é a propriedade característica para diferenciá-los.
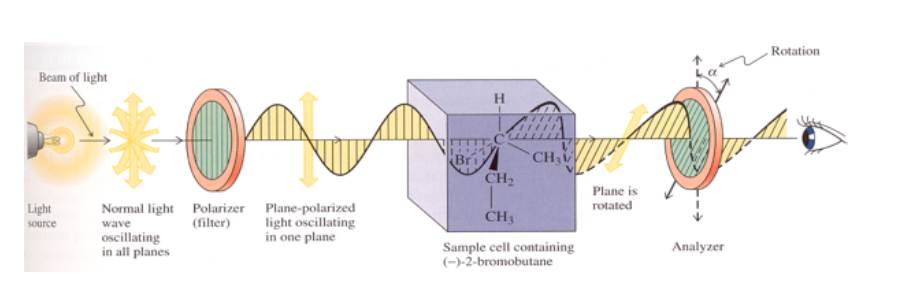
Se a substância não for opticamente ativa, nenhuma mudança no plano de vibração da luz polarizada emitida é observada.
Se a substância tiver atividade óptica, observa-se uma rotação de dois graus do plano de vibração da luz polarizada emitida.
Se a rotação do plano da luz for para a direita (no mesmo sentido horário), a substância é dextrorrotatória e o valor α recebe um sinal positivo.
Se a rotação for para a esquerda (sentido anti-horário), a substância é canhota e α recebe um sinal negativo.
A rotação específica de uma molécula opticamente ativa é uma constante física característica dessa molécula.

Rotação específica de alguns compostos quirais
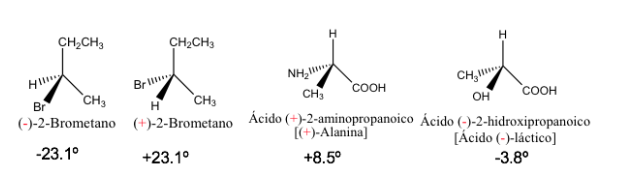
Os enantiômeros puros têm o mesmo valor de rotação específica, mas com o sinal oposto.
Portanto, a rotação óptica resultante de uma mistura 1:1 de enantiômeros é zero, ou seja, é opticamente inativa. Este tipo de mistura é chamado de racemato ou mistura racêmica .
A nomenclatura da configuração absoluta de um centro estereogênico é baseada nas mesmas regras de prioridade desenvolvidas por Cahn, Ingold e Prelog .
Essas regras permitem nomear e descrever o arranjo no espaço de substituintes em um centro estereogênico, independentemente do sinal da rotação óptica da molécula.
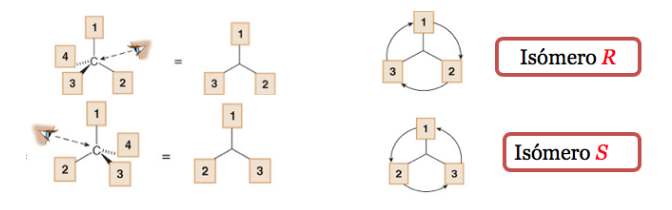
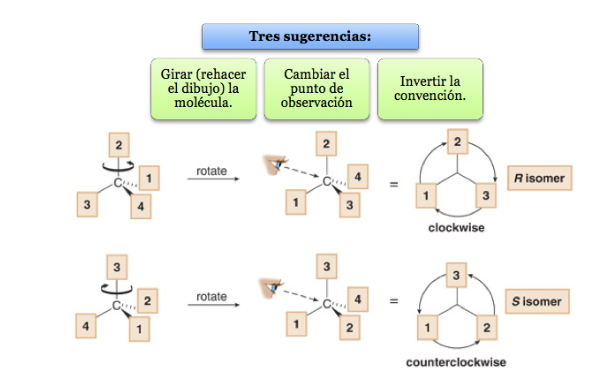
O substituinte de menor prioridade está localizado o mais longe possível do observador.
Se o passo de 1 para 2 para 3 for feito no sentido horário, o centro quiral é R (rectus, latim, direito).
Se o passo 1 a 2 a 3 for feito no sentido anti-horário, a configuração do centro quiral é chamada de S (sinistro, latim, esquerdo).
Na nomenclatura sistemática, R ou S são adicionados entre parênteses como um prefixo ao nome do composto quiral.
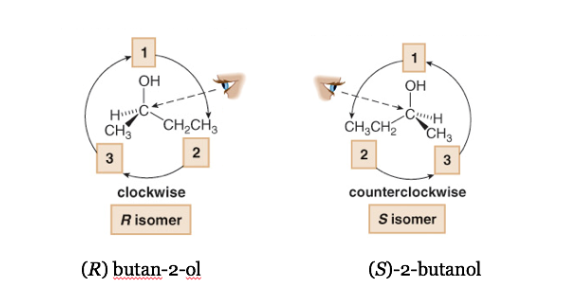
É importante lembrar que os símbolos R e S não apresentam nenhum tipo de correlação com o sinal de α .
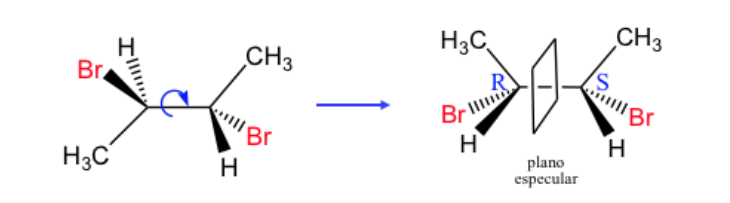
O que fazer quando uma molécula não está orientada de forma que o grupo de menor prioridade esteja longe?
Um composto com n centros estereogênicos tem no máximo 2 n estereoisômeros.
Exemplo:
Um composto com dois centros estereogênicos tem no máximo 4 estereoisômeros.
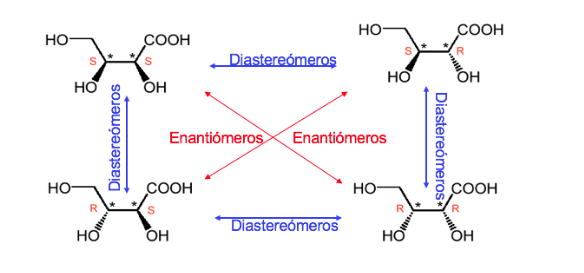
Qualquer estereoisômero cuja molécula não é quiral, apesar de ter centros estereogênicos, é chamado de forma ou composto meso .
Um composto com dois carbonos assimétricos é dominado como quando os dois carbonos têm a mesma configuração absoluta e diferente no caso contrário.
compostos eritro e treo
Quando dois carbonos têm pelo menos dois substituintes idênticos, pode-se usar a designação treo e eritro .
Um par de enantiômeros eritro é aquele em que grupos idênticos podem ser colocados na posição eclipsada.
Não há relação direta entre a nomenclatura R e S e a nomenclatura Erythro/Treo.
Quando dois carbonos têm três substituintes idênticos, a forma eritro é meso , pois apresenta um plano de simetria.
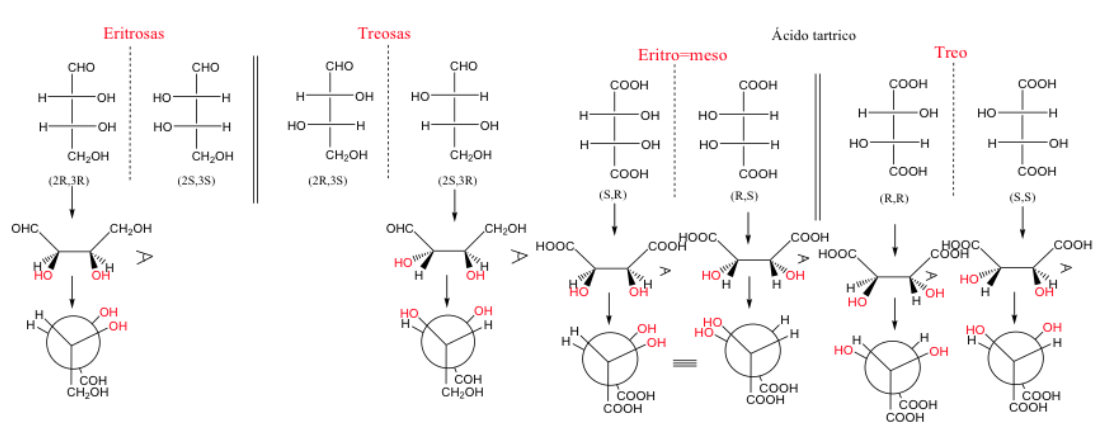
Uma molécula é chamada de eritro quando, em sua representação de Fischer, grupos iguais ou semelhantes estão do mesmo lado.
Uma molécula é três se esses grupos estiverem em lados opostos.
(geralmente usado para açúcares (osas))
É uma nomenclatura que antecede a nomenclatura R e S.
Um açúcar é denominado D quando, na projeção de Fischer (com o carbono mais oxidado localizado no topo), a hidroxila associada ao carbono assimétrico de maior número está à direita.
Seu enantiômero será chamado L e terá o equivalente OH à esquerda.
Glicose e frutose em suas formas naturais existem como D.
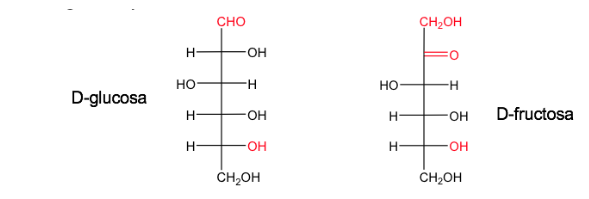
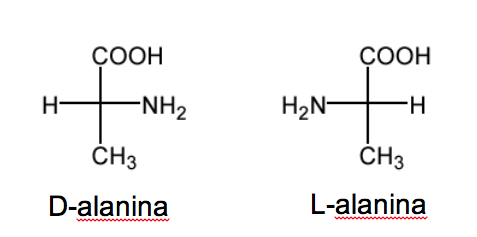
A nomenclatura D/L também é usada na série de aminoácidos.
RCH( NH2 )COOH.
Nos açúcares, esta nomenclatura depende da posição da hidroxila. Neste caso é a posição do grupo amino que define a nomenclatura. Quando na projeção de Fisher (com o carbono mais oxidado no topo) o grupo NH 2 está à direita, o etereoisômero é D e seu enantiômero é L .
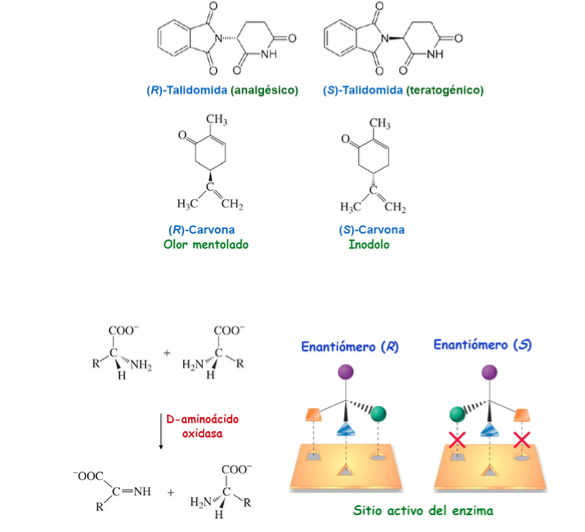
Importância da quiralidade
Fontes para ampliar a busca por conhecimento:
1) Juaristi E. “Introdução à estereoquímica e análise conformacional”. CINVESTAV, México, 1988 Juaristi E. . CINVESTAV, México, 1994.
2) Neil SI “Physical Organic Chemistry” Longman, Milão, 1995.
3) March J., “Química Orgânica Avançada” John Wiley & Sons, Nova York, 1992 4) Jones RAY “Química Orgânica Física e Mecanística”, 2º. Ed Cambridge University Press, Cambridge, 1984.
5) Woodward RB e Hoffmann R. “A conservação da simetria orbital”, Academic Press, Nova York, 1979.
6) Carpenter BK “Determinação de Mecanismos de Reação Orgânica”, John Wiley & Sons, Nova York, 1984 .

")
")
")
")
")
")
")

