SÍNTESIS ORGÁNICA
- Detalles
- Wilbertrivera
- SÍNTESIS ORGÁNICA
- Visto: 14564
Síntesis del poliaspartato (rootGrow)
1. Síntesis del poliaspartato (rootGrow)
2. Procedimientos de biosíntesis
Se afirma que la fórmula del rootGROW tiene relación con la del nutra-sweet
(aspartamo), un edulcorante poderoso, que está siendo retirado o ya lo fue del
mercado, debido a sus efectos nocivos en la salud de las personas.
El nutra-sweet, puede ser
sintetizado a partir de la fenilalanina, de acuerdo al esquema siguiente:

nutra sweet

A su vez, el ácido aspártico, base fundamental, par obtener el rootGrow,
que puede ser dextrógiro (D) o levógiro (L), se prepara de acuerdo a los
siguientes esquemas de síntesis:
Esquema A: (proceso de transaminación)

- Detalles
- Wilbertrivera
- SÍNTESIS ORGÁNICA
- Visto: 87564
SÍNTESIS DE LA PIRIDINA Y SUS DERIVADOS
|
La piridina es un líquido incoloro de olor desagradable, presenta en su estructura un heterociclo de seis eslabones, y tiene un carácter aromático; le sigue en importancia a la quinoleína debido a que se encuentra presente en numerosos alcaloides y fármacos de diversos usos. |
 |
|
|
Es muy soluble en agua, con un pKa 5.17 de punto de ebullición 115º C y es una base débil, pKa del amoníaco 9.2 de la piperidina 11.2 y un poco más fuerte que la anilina cuyo pKa es 4.6. Normalmente se lo extrae del alquitrán de hulla conjuntamente a las metilpiridinas, denominadas picolinas. |
 |
|
1. Reacciones químicas de la piridina
La piridina debido a su alta estabilidad no es afectada con los agentes oxidantes comunes, sin embargo se reduce con mayor facilidad que el benceno, para formar la correspondiente piperidina.
Por otro lado, se puede afirmar que la reactividad de la piridina es equivalente a la de una anillo bencénico disustituido con dos grupos nitro en posición para, razón por la cual no todas las sustituciones electrofílicas aromáticas que se dan en el benceno, ocurren en la piridina. Así por ejemplo no se producen la alquilación y acilación de Friedel Crafts, tampoco ocurre la nitración a temperaturas moderadas y de la halogenación la única reacción significativa es la bromación.
Las estructuras de resonancia de la piridina que reciben (o toman) un sustituyente B electrófilo y que a continuación se muestran, permiten la predicción de los lugares en los cuales se producirán las reacciones de sustitución electrofílica:

La inspección de las formas de resonancia cargadas sugiere que la densidad electrónica sobre los átomos de carbono alfa y gamma es especialmente baja; consiguientemente debe esperarse una sustitución en beta, además debido a que esta posición es la única en la que el estado de transición en la sustitución, no tiene una forma de resonancia con una carga sobre el nitrógeno trivalente.
ü Halogenación. La bromación es más estereoespecífica que la cloración, porque se puede obtener sólo el derivado monobromado en beta, separable de la dibromopiridina por destilación.

Sin embargo, también se pueden obtener derivados clorados, a partir de las alfa o gamma hidroxipiridinas

ü Sulfonación. La piridina puede sulfonarse a temperaturas altas utilizando como catalizador sulfato de vanadio, formando ácido sulfónico, con muy buenos rendimientos; importante intermedio para la síntesis de piridinas sustituidas.

2. Síntesis y reacciones de los derivados de la piridina. La química de los grupos sustituyentes en la piridina, depende de la posición de sustitución, habiéndose arribado luego de las observaciones empíricas a las siguientes generalizaciones:
a) Los grupos funcionales sustituidos en la posición beta (3 ó 5) de la piridina, presentan propiedades aromáticas características.
b) Los grupos funcionales ubicados en las posiciones alfa o gamma de la piridina (2,4 o 6) presentan reacciones características idénticas a los que están unidos al carbono alfa de los grupos carbonilo (por ejemplo acidez de los hidrógenos).
- Detalles
- Wilbertrivera
- SÍNTESIS ORGÁNICA
- Visto: 29918
Síntesis de compuestos 1,3 y 1,5-dicarbonílicos
Proponer un diseño de síntesis por el método de las desconexiones (método del sintón) a partir de materiales simples y asequibles, para las siguientes moléculas:
(Recuerde que si no es posible plantear una desconexión directa, será necesario recurrir a la estrategia de funcionalizar previamente la MOb, hasta llegar a un modelo de desconexión aplicable)

SOLUCIONES A LOS PROBLEMAS PROPUESTOS
La MOb 01, no presenta ninguna relación dioxigenada, aspecto que en algún sentido es una ventaja para el químico. Esto es, la estrategia a utilizarse, permitirá buscar relaciones dioxigenadas en la molécula precursora (equivalente sintético) con cierto grado de libertad, es decir, puede postularse según la estructura de la MOb, una gama de relaciones dicarbonílicas y/ó hidroxicarbonílicas, en posiciones relativas 1,2, 1,3, 1,4, 1,5 y/o 1,6 o sus variantes, como son los compuestos α, β-insaturado carbonílicos.
En esta oportunidad, se ejercitará las síntesis, recurriendo a las relaciones 1,3 y/o 1,5 dioxigenadas. De este modo se convertirá a la molécula objetivo y precursoras en estructuras desconectable según un modelo conocido y preestablecido.
Solución MOb 01
Análisis retrosintético: En la molécula que nos ocupa, se puede empezar por realizar un AGF, colocando un grupo C=O en la estructura de la molécula precursora, en posición tal, que permita luego realizar otro AGF, con un doble enlace ubicado en el carbono alfa y beta respecto al carbonilo, para proceder a desconectarlo.
La insaturación α, β, respecto del C=O, debe buscarse como el alqueno más sustituido de las alternativas que podrían existir. La presencia de un sustituyente en la posición beta al C=O, induce a pensar que el mismo pudo adicionarse como nucleófilo a un compuesto α, β insaturado carbonílico, según la reacción de adición conjugada de Michael. En base a estas consideraciones, puede postularse el siguiente análisis retrosintético, para la MOb 01:

Síntesis de la MOb 01: La condensación entre un aldehído y cetona enolizables, normalmente origina productos de autocondensación o de condensación cruzada. Esto se puede evitar, recurriendo a la estrategia de ejercer control, en el nucleófilo de la cetona, como puede verse en el esquema adjunto.
Por otro lado, cuando se requiere utilizar cetonas vinílicas como sustrato en la reacción de Michael, supondría la utilización del formaldehído. Lamentablemente este aldehído al ser muy reactivo en medio básico, tiende a originar reacciones de polimerización, que bajan drásticamente el rendimiento de la síntesis, por tal razón se combina adecuadamente las reacción de Mannich y la eliminación de Hofmann, para las cetonas vinílicas con rendimientos altos.
La eliminación de Hofmann, podría llevarse acabo en el mismo medio básico que se utiliza para la reacción de Michael, por lo que no es necesario aislar la vinilcetona.
Sin embargo, debido a la experiencia aún limitada en síntesis, se postulará la utilización de un óxido de plata en medio acuoso para lograr la eliminación de la amina y formación de la vinilcetona

- Detalles
- Wilbertrivera
- SÍNTESIS ORGÁNICA
- Visto: 34034
SÍNTESIS DE LA THORINA
|
Resumen: La síntesis de la Thorina, un reactivo cromógeno de amplio uso para el análisis espectrofotométrico de varios elementos, se encara dentro el paradigma de la retrosíntesis, lo que origina más de once rutas de síntesis diferentes, de las cuales se informa de dos de ellas. Los rendimientos y la identificación inequívoca de la thorina por IR y RMN, demuestran que los diseños de síntesis efectuados son los más adecuados y los espectros señalan del grado de pureza óptimo alcanzado en los procesos de recristalización de la Thorina sintetizada. |
|
|
|
|
|
|
|
El ácido 2-(3,6-disulfonato sódico-2-hidroxi-1-naftilazo) bencenarsónico, más conocido como thorín 1, thorina o simplemente torina, es ampliamente utilizado como reactivo cromógeno para el análisis espectrofotométrico de varios elemento, entre ellos el litio a 486 nm[1]. |
Thorina |
|

Las aplicaciones analíticas de la thorina, los parámetros de trabajo, sus limitaciones y perspectivas, son abordadas y explicadas con suficiente detalle, en las publicaciones especializadas de análisis químico como es el Analitical Chemistry. Un breve resumen, de los mismos, muestra las siguientes aplicaciones:
En medio ácido: se utiliza para la determinación cuantitativa de los elementos thorio (Th), zirconio (Zr), flúor (F), hafnio (Hf) y uranio (U).
En medio básico: Es utilizada la forma ácida disulfónica de la thorina, más conocida como Thorón, en la determinación cuantitativa del litio (Li).
En medio neutro: la thorina es utilizada como indicador para la determinación de iones sulfato (SO42- ) en soluciones acuosas.
La thorina también es utilizada para determinar el SO2 en el aire.
Problema de investigación.
Dada la demanda de este reactivo para diferentes determinaciones analíticas y particularmente para el análisis colorimétrico del litio en salmueras proveniente del Gran Salar de Uyuni y la poca oferta del mismo en el mercado nacional, surge la necesidad de sintetizar la thorina, a partir de materiales simples y asequibles en nuestro medio.
Con esta finalidad, las rutas de síntesis diseñadas, se lo encara desde el paradigma de la RETROSÍNTESIS, siendo aplicables los métodos de Las Hojas de Síntesis, Método de las desconexiones o del Sintón y el Método del Árbol de Síntesis.
En relación a la síntesis de la thorina, existe en la literatura una descripción breve y resumida de la síntesis del thorón[2], de acuerdo a la dirección propuesta por Kuznetsov, es decir, se indica que para la preparación del thorón, se requiere que el ácido o-aminofenilarsónico, diazocopule con la sal disódica del ácido 2-naptol-3,6-disulfónico (sal R) en medio ácido.
Diseño de síntesis para la Thorina.
El diseño se lo encara por el método de las desconexiones o del Sintón, que contempla dos etapas. La primera está relacionada con el análisis retrosintético y la segunda con la síntesis en dirección de lo que ocurre en el laboratorio, es decir a partir de los materiales de partida hasta arribar a la Molécula Objetivo (MOb).
a) Elementos estructurales y de reactividad a ser considerados, para el análisis retrosintético
La thorina es un compuesto típicamente azoico, por lo tanto un colorante. La preparación de estos compuestos comprenden generalmente dos reacciones fundamentales, que son: la diazoación y la diazocopulación (o simplemente copulación).
Por otro lado, todas las moléculas precursoras de copulación utilizadas para la formación de colorantes azoicos, poseen un carácter común, esto es: un átomo activo de hidrógeno ligado a un átomo de carbono.
Son de uso amplio como moléculas precursoras (sustratos) para la copulación, las siguientes: Compuestos que poseen hidroxilos fenólicos, como los fenoles y naftoles, Aminas aromáticas, Moléculas con grupo cetónico enolizables de carácter alifático y Moléculas heterocíclicas como el pirrol, indol, etc.
En relación a la reacción de copulación, se debe tener en cuenta ciertos principios heurísticos, a saber:
· Los fenoles se copulan más fácilmente que las aminas y los miembros de la serie del naftaleno más fácilmente que las del benceno.
· Los sustituyentes con efecto inductivo negativo (-I), como los halógenos, grupos nitro, sulfa, carboxilo y carbonilo retardan la copulación.
· Un grupo alquilo o alcoxi en posición orto o meta con respecto a un grupo amino estimula la copulación. Y si se hallan en las posiciones 2 y 5 con respecto al grupo amino, son copuladores especialmente buenos.
· En la serie del benceno, la copulación ocurre ordinariamente en la posición para con respecto al grupo hidroxilo (-OH) o amino (-NH2). Si la posición para esta ocupada, el enlace ocurre en la posición orto.
· En la serie del naftaleno, cuando el grupo hidroxilo amino está en la posición 2 (b), el reactivo se copula en la posición 1 (a). Si el grupo hidroxilo o amino está en la posición alfa, el enlace suele producirse en la posición 4; pero si la posición 3 o 6 están ocupados por un grupo sulfónico, la unión se efectúa en la posición beta.
· Cuando existen dos posiciones posibles de copulación, la posición del enlace es decidida frecuentemente por el pH del medio: La copulación se produce en orto con el grupo amino cuando se realiza en medio ácido y en orto con el grupo hidroxilo cuando se realiza en medio básico.
- Detalles
- Wilbertrivera
- SÍNTESIS ORGÁNICA
- Visto: 47442
Síntesis del Fenantreno
(Método del Árbol de Síntesis)
El Fenantreno es un hidrocarburo aromático policíclico, que contiene tres anillos bencénicos fusionados, razón por la cual se constituye en un isómero del antraceno.
|
|
ó |
|


Los métodos de síntesis tradicionales del Fenantreno, aquellos que conllevan la formación de los ciclos y su posterior “aromatización”, están vinculados a los propuestos por Haworth y a Bardhan – Sengupta (1932), como se verá a continuación.
Es posible también, proponer otros nuevos métodos, para el Fenantreno y en general para los compuestos aromáticos policíclicos, en base a la utilización adecuada de las reacciones de acoplamiento de Ullman, la reacción de Heck, la de Suzuki y la de MacMurry, así como las variantes y extensiones que presentan estas reacciones.
a) Síntesis de Haworth:
Este método, se basa en la reacción de acilación de Friedel – Crafts y presenta el inconveniente de que la ciclación final, para formar el tercer anillo fusionado al naftaleno, no es selectiva, debido a que también, es posible que el cierre se produzca en el otro carbono adyacente al grupo que contiene la función carboxílica y forme de este modo un isómero, que por las condiciones de reacción resulta ser el minoritario.
Método A:

Los materiales de partida, suelen ser el naftaleno y el anhídrido succínico. Para garantizar que la reacción de acilación con el anhídrido ocurra en el carbono 2 (beta) del naftaleno, es necesario realizar la reacción a una temperatura superior a los 60 ºC.
A temperatura ambiente, la posición de acilación será el carbono 1 (alfa), lo cual origina una variante al método, que sin embargo esencialmente son las mismas reacciones que ocurren y también existe la formación de otro isómero que es mucho menos significativo que en el primer caso. Por lo cual es preferiblemente utilizado como la reacción oficial de preparación del Fenantreno por el método de Howorth.
- Detalles
- Wilbertrivera
- SÍNTESIS ORGÁNICA
- Visto: 66008
SÍNTESIS DE BENZODIACEPINAS (BDZ)
Por. Wilbert Rivera Muñoz
Esta dirección de correo electrónico está siendo protegida contra los robots de spam. Necesita tener JavaScript habilitado para poder verlo.
Estas sustancias producen una gran variedad de efectos, que incluyen la sedación, el sueño, disminución de la ansiedad, relajación muscular esquelética, amnesia (olvido de situaciones a partir de la administración de la sustancia) y actividad anticonvulsiva.
A pesar de que todas las benzodiacepinas (BDZ) tienen acciones similares, existen diferencias en la selectividad con la que cada una de ellas produce alguno de los efectos anteriormente mencionados.
|
|
Su uso clínico varía según la estructura de la benzodiacepina, siendo la naturaleza de los grupos R en la posición 1, 2, 5 y 7, las variables fundamentales que caracterizan al fármaco en cuestión. Sin embargo la profundidad de los efectos, depende de la dosis, a medida que ésta aumenta, van apareciendo signos de sedación, sueño y hasta coma. La primera molécula preparada de este tipo fue el Clordiacepóxido en 1934 por Henrik Leo Stembach, pero debido a la segunda Guerra Mundial, recién fue introducida en la práctica médica en los años cincuenta. |
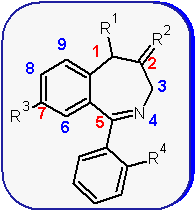
En los años 60 el mismo Stembach, inició en la empresa farmacéutica Hofmann-La Roche, el preparado de las benzodiacepinas, originando así el vertiginoso desarrollo de los considerados “tranquilizantes menores”.
Desde un punto de vista químico, las benzodiacepinas se pueden dividir en dos grupos: las 1,4-benzodiacepinas simples (como el diacepam) y las 1,4-benzodiacepinas heterocíclicas (como el alprazolam). En la actualidad, se han sintetizado una gran cantidad de BDZ, de los cuales se han probado un buen número y tan sólo un poco de ellas se encuentra en uso clínico.
Su extraordinaria popularidad se basa en su capacidad para reducir la ansiedad (definida como angustia en ausencia de un objeto real que la produzca directamente) sin interferir demasiado con otras funciones ligadas al estado de consciencia.
Sin embargo, todas las BDZ tienen efectos sedantes e hipnóticos, propiedades que han hecho que se las utilice ampliamente en lugar de los barbitúricos. Varias de ellas también se usan, por periodos breves, como relajantes musculares.

")
")
")
")
")
")
")

